● INTRODUCTION
Older literature suggested that genetic diseases contributed minimally to the origin of congenital heart disease (CHD), with the majority of defects thought to result from multifactorial causes. Current knowledge suggests that the genetic contribution to CHD has been significantly underestimated in the past. Recent advances in genetic research have added significantly to the knowledge of early cardiac development and to the etiology of CHD (1) (see Chapter 3). The field of human cardiovascular genetics is progressing at a rapid pace, and the availability of novel genetic tests for various forms of cardiac abnormalities are regularly being introduced to clinical medicine (2). The recent introduction of noninvasive prenatal screening is an example of how genetic innovations are having substantial impact on clinical care. In this chapter, we will present the typical genetic diseases related to cardiac anomalies.
● GENETIC TESTING
Chromosomal Analysis
The chromosomal constitution of a cell is referred to as its karyotype and there are international standards for analyzing and reporting chromosomal finding. Chromosomal analysis is done on cell cultures that are treated to arrest growth in the metaphase when the chromosomes are visible. Special banding techniques (usually G-banding) are used to identify individual chromosomes by their specific pattern of light and dark bands. The analysis is performed under the microscope and the chromosomes are classified into seven groups, A to G, based on their length and centromere position. Nomenclature designates the centromere as “cen” and the telomere (terminal structure) as “ter.” The short arm of each chromosome is designated as “p” (petit) and the long arm “q” (queue). Each arm is subdivided into a number of bands and sub-bands.
This traditional karyotyping technique identifies the majority (>75%) of clinically significant chromosome abnormalities including trisomies 21, 18, and 13, triploidy, and aneuploidies involving the sex chromosomes, such as monosomy X (Turner syndrome) and XXY (Klinefelter syndrome). Some large balanced or unbalanced translocations can also be found in addition to rare mosaic trisomies and marker chromosomes. Large chromosomal deletions can also be identified, such as the majority of deletions 4p- (Wolf–Hirschhorn syndrome). Small deletions, termed microdeletions, such as 22q11 (DiGeorge syndrome), are typically too small to be identified by this method. Microdeletions can be detected by the use of selective fluorescence in situ hybridization (FISH) when such condition is suspected (e.g., FISH for deletion 22q11 in conotruncal anomalies) or by examining the complete chromosome using comparative genomic hybridization (CGH) (microarray) (discussed later).
FISH Technique
FISH is a cytogenetic technique using specific fluorescent probes, which are applied to detect and localize the presence or absence of specific DNA regions on chromosomes. FISH uses a single DNA strand, called probe, corresponding to a specific locus and only binding to the corresponding complementary part on the chromosome. Fluorescence with various colors is used to enable visualization under the fluorescence microscope. FISH can be directly used on cells during cell division and as such it is called in situ interphase FISH, typically applied antenatally for a rapid diagnosis of trisomies. FISH technique used for the identification of microdeletions is performed directly on interphase chromosomes with the addition of the FISH probes. In general, two probes are used. The first probe (green) is a control probe used for the identification of both copies of the target chromosome. The second probe (red-magenta) hybridizes to the sequence of the region of interest on the target chromosome. In the presence of a deletion, which is typically on one of the paired chromosomes, a lack of the red signal is noted, as the probe cannot bind to the target region on the chromosome. When a specific cardiac anomaly is diagnosed in the fetus and an invasive procedure is performed, it has become customary to offer FISH for deletion 22q11.2 in addition to chromosome karyotyping.
Array Comparative Genomic Hybridization
Array CGH, or microarray, is even more sensitive than the two previous techniques and compares the patient’s DNA in all chromosomes with a control DNA sample for the identification of variances between the two sets. Imbalances in the patient’s DNA, such as deletions and duplications, can be identified with this technique. Instead of examining one deletion, such as with FISH, all possible regions of the chromosomes are examined for deletions and duplications and other imbalances. Explanation of the technical aspect of CGH is beyond the scope of this book, but it is important to state that CGH detects all DNA imbalances in chromosomes, some of which may have unclear clinical significance. This new microarray technique has become popular in the last years despite its cost and limitations. Some centers offer CGH as a first-line genetic testing after chorionic villus sampling or amniocentesis, while others restrict its use when DNA imbalance is suspected or as a second-line test following normal karyotypic analysis. In a recent meta-analysis (3), it was shown that for fetal heart defects CGH detected additional 7% of chromosomal abnormalities after excluding aneuploidies and deletion 22q11.2. With this finding, it is reasonable to discuss the option for CGH with the patient when fetal malformations and particularly CHD is diagnosed. The availability and cost of CGH should also factor into this decision. As more information accumulates on the association of CHD with chromosomal imbalances, the role of CGH in the diagnosis and management of CHD will be more clarified.
Noninvasive Prenatal Testing
Noninvasive prenatal testing (NIPT) is a relatively new genetic testing that is offered as a screening test in the first and second trimesters of pregnancy for trisomies 21, 13, and 18, monosomy X, and sex chromosomes abnormalities. The test is based on the presence of fetal cell-free DNA (cfDNA) in the maternal circulation primarily from placental cell apoptosis (4). Placental cell apoptosis releases into the maternal circulation small DNA fragments that can be detected from about 4 to 7 weeks’ gestation (5). It is estimated that about 2% to 20% of circulating cfDNA in the maternal circulation is fetal in origin (5). The half-life of cfDNA is short and is typically undetectable within hours after delivery (6). Details of the technical aspect of NIPT are beyond the scope of this book but the various tests that are clinically available are based on the isolation and counting of cfDNA using sequencing methods.
NIPT has very good performance with regards to screening for trisomy 21. In published studies, the detection rate for trisomy 21 is at 99% for a false-positive rate of 0.16% (7). Detection rate for trisomy 18 is at 97% for a false-positive rate of 0.15% (7). To date, NIPT has been recommended as a screening test for the high-risk population. Given a very small false-positive rate, incorporating NIPT for trisomy 21 screening in the high-risk population reduces the need for unnecessary invasive testing.
It should be emphasized that NIPT is a screening and not a diagnostic test and thus caution should be used when NIPT is incorporated in the genetic evaluation of CHD. Given a relatively high association of CHD with chromosomal imbalance, the significance of a normal NIPT result in the setting of CHD should be explained to the patient and further invasive diagnostic testing should be recommended. Undoubtedly, NIPT technology will expand over the next few years to allow for screening of chromosomal deletions and duplications. As a new and emerging technology, the role of NIPT in CHD should be carefully evaluated before a change in management guidelines is adopted.
● SONOGRAPHIC EVALUATION OF THE FETUS WITH CHD
The antenatal detection of CHD in a fetus necessitates a detailed ultrasound evaluation given a high association with extracardiac anomalies, ranging between 30% and 50% in some series. Establishing whether the CHD is isolated or part of a genetic syndrome is essential for patient counseling and for the assessment of long-term prognosis. On rare occasions, the type of CHD by itself provides enough information about associations, or lack thereof, to allow for patient counseling. For instance, this can be true for cardiac rhabdomyomas and their associations with tuberous sclerosis complex (TSC) or the commonly isolated simple transposition of the great arteries. For most CHD, however, there is a wide range of possible associations and detailed fetal evaluation is therefore warranted. Usually, the first step is to seek for the presence of additional soft markers and anomalies that suggest the presence of one of the common numerical chromosomal anomalies that can be detected by karyotyping. Furthermore, a detailed sonographic evaluation of the fetus is essential with a thorough search for markers of associated genetic syndromes. This can only be achieved if the examiner is aware of various associations and the phenotypic aspects of various syndromes. In the presence of a normal fetal karyotype, a discussion on the benefit of additional genetic testing should be undertaken with the patient. Tables on various associations of CHD with genetic abnormalities are available and have traditionally guided clinical management in such cases. The presence of subtle signs on ultrasound can point the examiner to a genetic association that may not be clearly visible otherwise. Tetralogy of Fallot is a typical example, where it can be isolated, but also is typically associated with trisomies 21 and 18, deletion 22q11.2, Alagille syndrome, CHARGE syndrome, and others. Another example is the presence of atrioventricular septal defect (AVSD), since it is suggested that more than 50% of AVSDs are associated with either trisomy 21 or 18. AVSD can also be part of heterotaxy syndrome (see Chapter 30), either isolated or in the context of primary ciliary dyskinesia. AVSDs were also observed in deletion 22q11.2 and other deletions as well as in CHARGE syndrome (13%) as has been recently reported (8).
● CHD AND NUMERICAL CHROMOSOMAL ANOMALIES
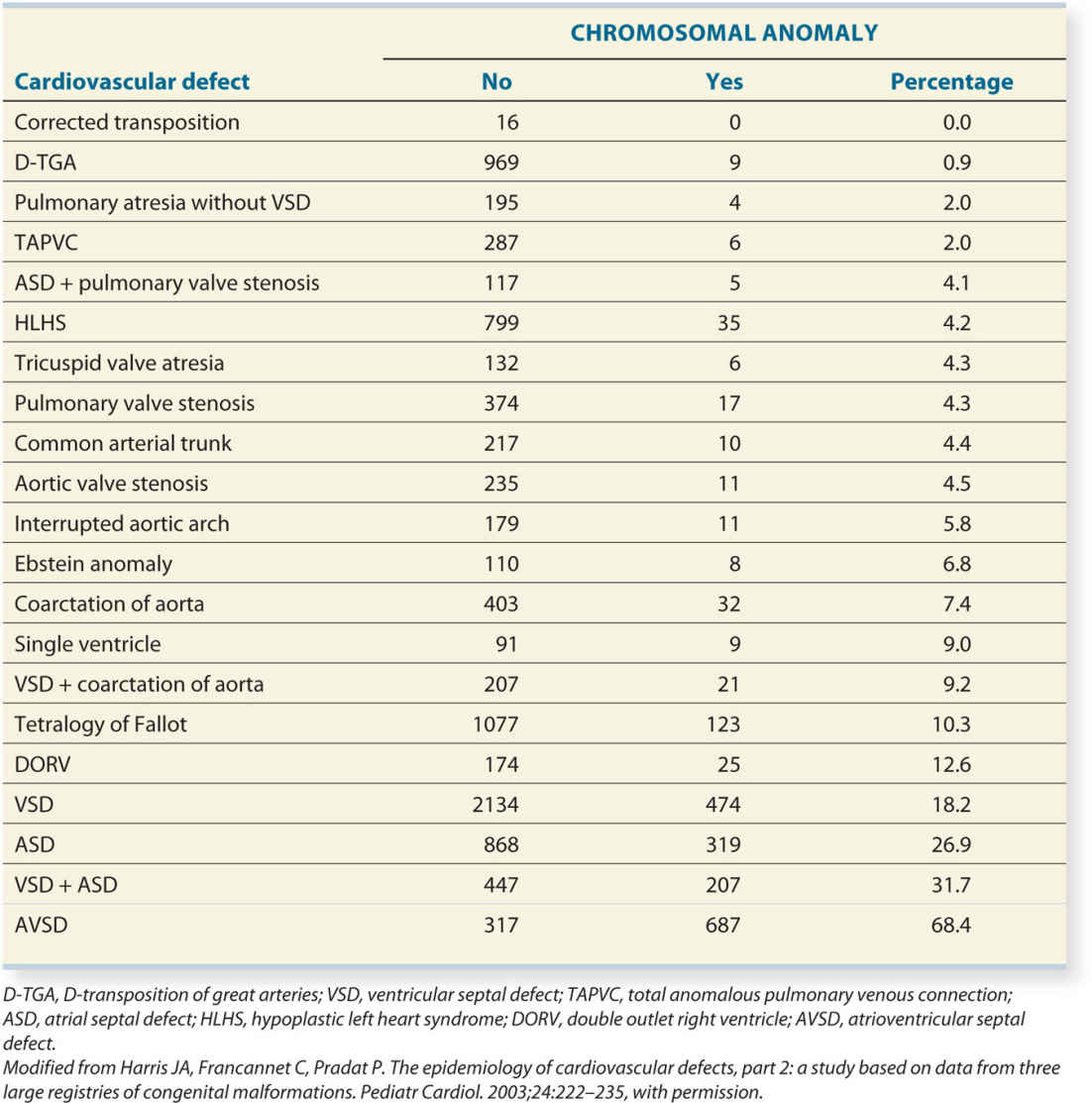
The frequency of chromosomal abnormalities in infants with congenital heart defects has been estimated as 5% to 15% from postnatal data (9–11). In a population-based case-control study of 2,102 live-born infants, ascertained by their cardiovascular malformations, chromosomal abnormalities were found in 13% (10). In this study, Down syndrome occurred in 10.4% of infants with cardiovascular malformations, with the other trisomies each occurring in less than 1% of cases (10). Similar data were reported from three large registries of congenital malformations, involving 1.27 million births (11). The frequency of abnormal karyotype in fetuses with cardiac defects is higher and has been reported in the range of 30% to 40% by several studies (12–14). This higher rate of chromosomal abnormalities in fetuses with cardiac defects, when compared to their live-born counterparts, is mainly due to an increased prenatal mortality in fetuses with aneuploidy, which has been estimated at 30% for trisomy 21, 42% for trisomy 13, 68% for trisomy 18, and 75% for Turner syndrome (15). Not only is the association of congenital cardiac defects and chromosomal abnormalities lower in live-born infants than that in fetuses, but also is the distribution of chromosomal abnormalities more skewed toward Down syndrome in the neonatal population (10, 11), again probably due to the high prenatal mortality of trisomies 18 and 13 and monosomy X.
Certain specific cardiac diagnoses are more commonly associated with chromosomal abnormalities than others. Prenatal and postnatal studies are concordant with regard to the specific cardiac diagnoses that are more likely to be associated with chromosomal abnormalities. In general, malformations of the right side of the heart are less commonly associated with karyotype abnormalities. Specific cardiac diagnoses, such as transposition of the great vessels and heterotaxy syndromes, are not usually associated with chromosomal abnormalities. AVSD, ventricular (perimembranous) and atrial septal defects, tetralogy of Fallot, double outlet right ventricle, and hypoplastic left heart syndrome, on the other hand, are more commonly associated with chromosomal abnormalities in the fetus and newborn. Table 4.1 lists specific cardiac diagnoses in infants with noncomplex cardiovascular defects from three large registries (11) and the corresponding incidence of associated numerical chromosomal abnormalities.
The majority of fetuses with cardiac defects and chromosomal abnormalities have other associated extracardiac abnormalities, in the order of 50% to 70% (12, 14). The distribution of extracardiac abnormalities usually follows the typical pattern noted within each chromosomal syndrome with no predominance of any specific abnormality. In the fetus with an apparently isolated cardiac abnormality, the incidence of chromosomal abnormalities is still significantly increased (15%–30%), and thus appropriate genetic counseling is warranted (12, 14).
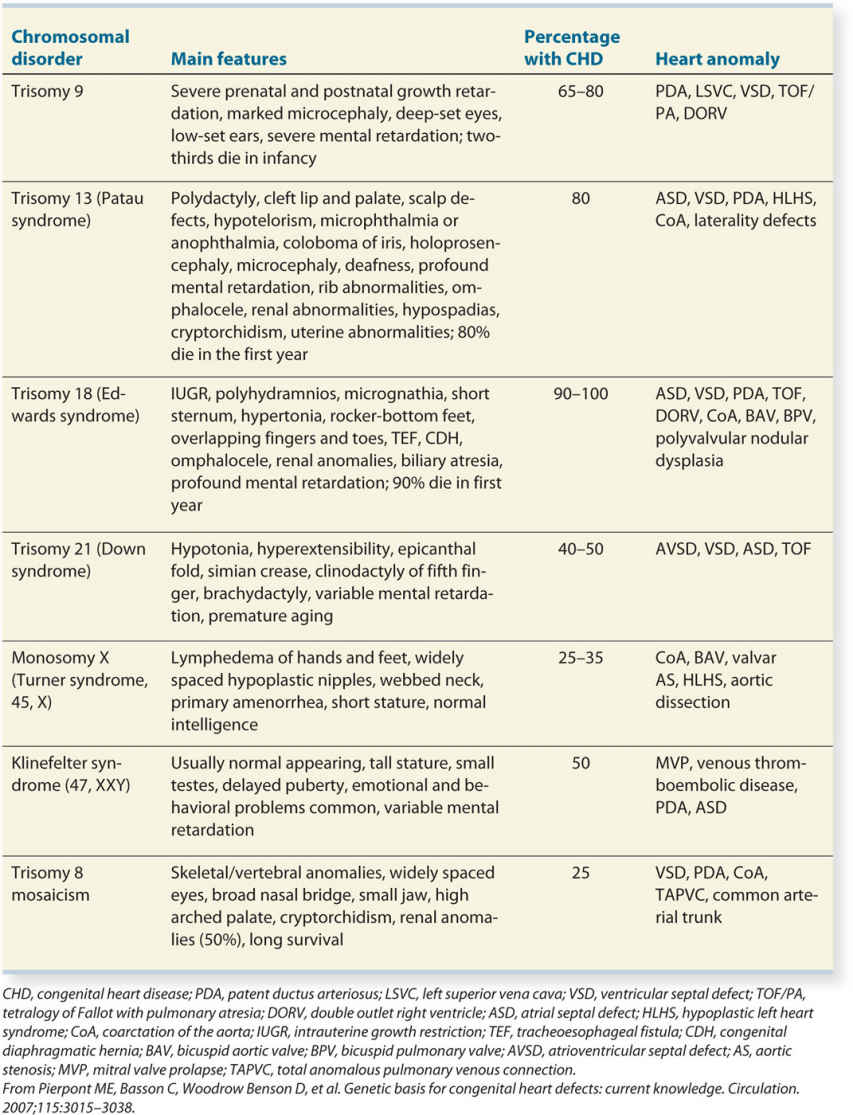
When the diagnosis of a chromosomal abnormality is made in a fetus, an echocardiogram is indicated in view of the common association of cardiac malformations with karyotype abnormalities. Data obtained from postnatal studies suggest that the incidence of cardiac defects is 40% to 50% in trisomy 21, 25% to 35% in Turner syndrome, and more than 80% in trisomies 13 and 18 (2, 16). Cardiac defects tend to be specific to the type of chromosomal abnormality. Table 4.2 lists the most common numerical chromosomal abnormalities and their associated congenital cardiac defects.
● CHD AND CHROMOSOMAL DELETION SYNDROMES
DiGeorge Syndrome (Deletion 22q11.2)
Definition of Disease
DiGeorge syndrome, also known as monosomy 22q11.2 deletion syndrome, velocardiofacial syndrome, or CATCH-22, is the most common deletion in humans and is the second most common chromosomal anomaly in infants with CHD (second to trisomy 21). It has an estimated prevalence of 1:2,000 to 1:4,000 live births (17). DiGeorge syndrome results from a deletion in the region 11 on long arm of chromosome 22. The acronym CATCH-22 was used to describe features of DiGeorge syndrome to include cardiac anomalies (C), abnormal facies (A), thymus hypoplasia (T), cleft palate (C), hypocalcemia (H), and the microdeletion on chromosome 22. Phenotypic abnormalities include cardiac outflow tract abnormalities in combination with thymus hypoplasia or aplasia, cleft palate, velopharyngeal insufficiency, and dysmorphic facial features (18). The clinical feature of the 22q11.2 deletion is, however, highly variable. Cardiovascular anomalies, which can be present in up to 85% of cases, immunodeficiency, and speech delay appear to be the most frequent phenotypic manifestations (18, 19). Other abnormalities include neonatal hypocalcemia due to parathyroid hypoplasia, feeding and behavioral disorders, learning disabilities, and cleft anomalies (2). Disorders of the skeleton can affect the limbs and the spine (20). Mental disorders are found in 30% of the adults with this deletion (17).
Genetic Diagnosis
Diagnosis of this microdeletion can be achieved by FISH technique or with microarray analysis. FISH for 22q11.2 deletion must be specifically requested in addition to routine karyotyping when looking for this deletion. The deleted region in this anomaly includes more than 40 genes, but one of these genes, namely the TBX1 gene, seems to be responsible for the cardiac and some other additional related anomalies. Mutation in the TBX1 gene can explain the rare cases of a clinical suspicion of deletion 22q11.2, but with no microdeletion of chromosome 22. In an affected fetus or infant, the parental examination reveals in approximately 6% an affected parent with subtle signs of this syndrome with a 50% transmission to future offspring (19).
Cardiac Findings
Cardiac anomalies that are found in deletion 22q11.2 syndrome primarily include conotruncal anomalies, such as interrupted aortic arch, common arterial trunk, absent pulmonary valve syndrome, pulmonary atresia with ventricular septal defect, tetralogy of Fallot, and conoventricular septal defects (21–23) (see examples in corresponding chapters). The presence of a right aortic arch either isolated or in combination with a cardiac anomaly increases the risk of deletion 22q11.2. Other cardiac anomalies can be found but in less than 5% of cases. Table 4.3 estimates the 22q11.2 deletion with various types of CHD.
Main Extracardiac Findings
Once a cardiac anomaly is diagnosed prenatally, the additional demonstration of a hypoplastic or absent fetal thymus on an ultrasound examination increases the risk of an association with a deletion 22q11.2 (22, 23). The thymus is demonstrated in a transverse plane of the chest at the level of the upper sternum (three-vessel-trachea view) anterior to the three vessels (see Chapter 9). Thymic aplasia or hypoplasia may be suspected at this plane level. Note that the presence of a thymus on ultrasound does not rule out a 22q11.2 deletion (23). Typical facies with bulbous nose, ear anomalies, polyhydramnios, skeletal findings including polydactyly, club foot, spinal anomalies (hemivertebra, spina bifida), renal anomalies, and others were reported (20, 24).
Prenatal Reports
Numerous reports on prenatal cardiac defects and features of deletion 22q11.2 in the fetus are available. These reports highlight the cardiac defects found and the role of evaluating the thymus when deletion 22q11.2 is suspected. Two recent studies report on the wide spectrum of anomalies in the fetus with DiGeorge on ultrasound and autopsy (20, 24).
Estimated 22q11.2 Deletion Frequency in Congenital Heart Defects |
Cardiac defect | Estimated deletion frequency (%) |
Interrupted aortic arch | 50–89 |
VSDs With normal aortic archa With aortic arch anomalyb | 10 3 45 |
Common arterial trunk | 34–41 |
Tetralogy of Fallot (including pulmonary atresia with VSD and absent pulmonary valve syndrome) | 10–40 |
Isolated aortic arch anomalies | 24 |
Double outlet right ventricle | <5 |
Transposition of the great arteries | <1 |
aLeft-sided aortic arch with normal branching pattern.
bIncludes right aortic arch and/or abnormal branching pattern, cervical location, and/or discontinuous branch pulmonary arteries.
Adapted from Pierpont ME, Basson C, Woodrow Benson D, et al. Genetic basis for congenital heart defects: current knowledge. Circulation. 2007;115:3015–3038; and Chaoui R, Kalache KD, Heling KS, et al. Absent or hypoplastic thymus on ultrasound: a marker for deletion 22q11 in fetal cardiac defects. Ultrasound Obstet Gynecol. 2002;20:546–552, with permission.
VSD, ventricular septal defect.
Williams–Beuren Syndrome
Definition of Disease
Also called Williams syndrome, the disease is a multisystem disorder and is described by characteristic facies, called “elfin facies,” and the presence of cardiac anomalies, infantile hypercalcemia, skeletal anomalies, renal anomalies, and cognitive abnormalities (25) with mental retardation. In general, diagnosis is achieved at various ages in childhood. The disease occurs with an incidence of 1:20,000 live births.
Genetic Diagnosis
The disease is caused by a microdeletion at chromosome 7q11.23, which includes the elastin gene, ELN. This can be confirmed by targeted FISH technique or CGH array diagnosis (21).
Cardiac Findings
Characteristically, children with Williams syndrome have supravalvular aortic and pulmonary stenosis with narrowing in the vessel wall in the region distal to the valve. This condition cannot be routinely diagnosed in the second trimester of pregnancy (Table 4.4). In addition, an aortic coarctation is often present and occasionally peripheral pulmonary stenosis.
Main Extracardiac Findings
In addition to the cardiac defect, mild growth restriction can be present, including a short femur as reported in one case (26). Due to elastin gene involvement, cardiac and extracardiac calcifications can be found in children with Williams syndrome. No other characteristic signs are found on prenatal ultrasound.
Prenatal Reports
Williams syndrome is often missed in the fetus due to the noncharacteristic and minimal cardiac findings (26). The authors routinely offer a targeted FISH examination for Williams syndrome in cases with fetal left ventricular outflow tract obstruction (aortic coarctation, aortic stenosis, and hypoplastic left heart syndrome) and despite this approach, we have not diagnosed a single case of Williams syndrome prenatally over several years. Improved prenatal detection can be achieved by the common use of microarray analysis or targeted panels for genetic syndromes with invasive diagnostic procedures. The diagnosis of Williams syndrome was detected prenatally by performing comprehensive genetic testing at 30 weeks’ gestation for fetal growth restriction (27). The targeted ultrasound showed the poststenotic narrow ascending aorta as a sign of poststenotic aortic stenosis (27).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


