Transplant Immunology and Clinical Immunosuppression
Anne C. Fischer
Paul M. Colombani
Division of Pediatric Surgery, Johns Hopkins University, Johns Hopkins Hospital, Baltimore, Maryland 21287.
Division of Pediatric Surgery, Johns Hopkins University, Johns Hopkins Hospital, Baltimore, Maryland 21287.
Immunology is a relatively new scientific field whose origins are traced to 1796. Edward Jenner’s discovery that prior exposure to cowpox or vaccinia protected against developing human smallpox formed the theoretical basis of vaccination. Thus, exposure to weakened or attenuated strains of pathogens could prevent acquisition of disease. Jenner’s discovery of protective immunity even predated Koch’s unraveling of the microbiologic basis of infectious diseases. Heretofore, the influence of Hippocrates and Galen dominated the prevailing theories that illnesses resulted from the imbalance of “humors,” or body fluids. Therefore, Pasteur’s “germ theory of disease” was remarkably novel, as was his broader application of vaccination to any pathogen. This area of work on protective immunity culminated in 1901 in Emil von Behring’s Nobel Prize, demonstrating that the serum of vaccinated individuals contains humoral components capable of binding relevant pathogens. Thus, the science behind a specific humoral immune response such as an antibody to a particular antigen, or protective immunity, was born. At the same time, Elie Metchnikoff, a Russian immunologist, identified the first cellular components of the immune system: phagocytic cells. The science of immunology was then divided between opposing schools of thought—the humoralists and the cellularists—with little appreciation as yet of the redundancy of the immune response. Although the earliest discovers laid down the theoretical basis of immunology, it was not until the actual appreciation of the multiple components of the immune system, both humoral and cellular, was transplantation possible. It was actually the study of mouse skin allograft rejections that identified the major histocompatibility complex (MHC) molecules critical for self : nonself recognition. In fact, the immunobiology of transplantation has elucidated our current understanding of immunology. The major impediment to transplantation is fundamentally the host’s adaptive immune response to the graft’s foreign proteins, including alloantigens and ABO/Rh blood group antigens (Fig. 45-1). To appreciate the complexity of the immune response to foreign alloantigens, the components of the immune system are reviewed first, followed by the strategies of immunosuppressive regimens intended to induce long-term graft tolerance. Not until the 1950s was the technical skill of vascular anastomoses applied to achieve the first living related kidney transplant between identical twins (1). Technical mastery still needed to be matched by advances in immunosuppression, specifically targeted to the alloantigen effector arm, prior to ensuring successful solid organ transplantation. Both of these advances further required a unified network of transplantation centers organized to ensure maximal tissue compatibility and recipient suitability.
IMMUNOLOGIC ONTOGENY
All cells from the immune system are derived from a single pluripotent hematopoietic stem cell, influenced by various growth factors. Hematopoiesis is first detected in the third week of gestation, and after 5 weeks, the yolk sac hematopoietic activity declines from a maximal level to being undetectable by 60 days (2,3). The fetal liver then becomes the site of hematopoiesis during gestation until the last 2 months (4). Fetal stem cells possess both the capacity for self-renewal and the ability to differentiate into multiple hematopoietic lineages (5,6,7). During this prenatal period the site of hematopoiesis migrates to the bone marrow, which becomes the repository of hematopoiesis throughout adult life. Extramedullary hematopoiesis can occur postnatally in the spleen or liver during periods of severe stress. The first stage of differentiation of the pluripotent stem cell is between lymphoid and myeloid lineages. Cells from a particular hematopoietic lineage or stage of differentiation are phenotypically classified by monoclonal antibodies to their cell surface differentiation antigens. The cluster of differentiation antigens refers to clusters of antibodies binding to specific cell markers, characterizing the lineage and differentiation of all cells in the immune system (Table 45-1). The lymphoid lineage includes B lymphocytes (or bone marrow-derived lymphocytes), T lymphocytes (thymus-derived lymphocytes), and natural killer (NK) cells. The myeloid lineage encompasses erythrocytes, monocytes, macrophages, eosinophils, basophils, neutrophils, mast cells, platelets, and dendritic cells. Early myeloid and lymphoid development occurs initially in bone marrow, with final maturation in the peripheral blood and lymphatic organs upon antigenic exposure. T lymphocytes also arise from the bone marrow precursor cell, and migrate to the thymus prenatally and postnatally for selection and final T-lymphocyte maturation.
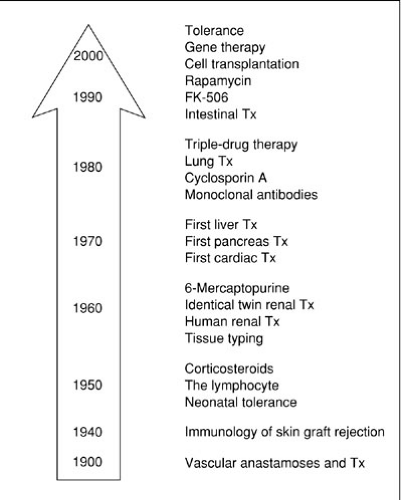 FIGURE 45-1. The explosion in knowledge due to basic science research in diverse fields has led to an equivalent increase in clinical applications. Tx, transplantation. (Adapted from Conrad BG, Ildstad ST. Chimerism and tolerance: the myth becomes a reality. In: Sher LS, Makowka L, eds. Intra-abdominal organ transplantation 2000. Austin, Tx: RG Landes, 1994:82.) |
TABLE 45-1 CD markers. | ||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
OVERVIEW OF THE IMMUNE SYSTEM
The immune system is divided primarily into humoral and cellular responses, as well as specific and nonspecific responses. Nonspecific or nonimmune host defenses include (1) the complement cascade and the alternative or properdin pathway in the humoral arm, and (2) nonspecific effector cells such as polymorphonucleocytes (PMNs) and monocyte-macrophages in the cellular arm. Chemotactic factors released at sites of infection cause the influx of PMNs, then serum opsonizing factors target the pathogen for engulfment, phagocytosis, and peroxidation by oxygen-free radicals. Specific immune defenses mount an immune response to foreign alloantigens, key targets for transplantation rejection. B lymphocytes and antibody production are the main humoral arm of specific immunity, whereas T lymphocytes constitute the cellular arm. T cells differentiate into effector cells and memory cells, produce cytokines to clonally expand T cells and augment other cells of the immune response, and directly destroy cells seen as foreign.
There is a complex orchestrated series of events that constitute a specific allograft immune response. First, the initial interaction between the graft’s alloantigens and the host antigen-presenting cells (APCs), including B cells, dendritic cells, macrophages, and monocytes, triggers the afferent limb of allorecognition (8). Foreign alloantigen is phagocytosed by APCs and digested into small epitopes approximately 5 to 15 amino acids long (9,10). These epitopes are displayed on the surface of the APC to activate T or B lymphocytes, causing allosensitization. Only those
lymphocytes with a specific receptor to the alloantigen, such as sensitized lymphocytes, can recognize the antigen and undergo activation. Once the afferent limb of the immune response, allosensitization, occurs, then alloantigen recognition by sensitized cells or antibodies elicits the cellular effector response, constituting the efferent limb of the immune response (11). The efferent limb of allograft rejection is initiated both by antibody-producing plasma cells specific to the alloantigen and by T cells eliciting a cellular inflammatory response to the graft. Allosensitized T cells are the primary effectors of allograft rejection, although NK cells and macrophages may play a role. The period of time to rejection is dependent on the state of sensitization of host immune cells, the type of allograft (skin vs. vascularized organs), and the immunologic disparity between donor and recipient (12,13). In essence, the extent of an immune response depends on multiple factors, the degree of allosensitization, and the robustness of the recipient’s immune system.
lymphocytes with a specific receptor to the alloantigen, such as sensitized lymphocytes, can recognize the antigen and undergo activation. Once the afferent limb of the immune response, allosensitization, occurs, then alloantigen recognition by sensitized cells or antibodies elicits the cellular effector response, constituting the efferent limb of the immune response (11). The efferent limb of allograft rejection is initiated both by antibody-producing plasma cells specific to the alloantigen and by T cells eliciting a cellular inflammatory response to the graft. Allosensitized T cells are the primary effectors of allograft rejection, although NK cells and macrophages may play a role. The period of time to rejection is dependent on the state of sensitization of host immune cells, the type of allograft (skin vs. vascularized organs), and the immunologic disparity between donor and recipient (12,13). In essence, the extent of an immune response depends on multiple factors, the degree of allosensitization, and the robustness of the recipient’s immune system.
THE MAJOR HISTOCOMPATIBILITY COMPLEX
The MHC is a highly polymorphic chromosomal region that encodes for cell surface glycoproteins responsible for alloreactivity and rejection (14). The primary function of these glycoproteins is to bind peptides and present them properly for recognition by T cells. The MHC corollary in humans is the human leukocyte antigen (HLA) complex, which represents the most important determinants of alloreactivity and thus refers to major antigens. Those determinants encoded outside the MHC still can contribute to long-term rejection, but are less antigenic and notably referred to as minor antigens.
The MHC is located on the short arm of chromosome 6 in humans (called the HLA complex) and contains more than 200 genes and 4 × 106 base pairs reflecting significant nucleotide sequence diversity. There are three basic loci encoding for HLA class I, II, and III proteins (Fig. 45-2). Despite a large polygene complex, only the class I and II molecules determine rejection and are critical for transplantation. Both HLA class I proteins, which are constitutively expressed on the surface of all nucleated cells (not erythrocytes) and HLA class II proteins, primarily expressed on APCs, are involved in antigen presentation. Class I expression can be upregulated by cytokines such as TNF-α and α-IFN (tumor necrosis factor and interferon, respectively); likewise, class II expression is upregulated by γ-IFN. The three most common class I genes are HLA-A, -B, and -C, and all are involved in antigen presentation. The others, called class IB genes, are HLA-E, -F, -G, and -H, which are class I-like and have various other functions (15). The HLA class II region encodes genes called HLA-DR, -DP, and -DQ. The HLA-DR locus can contain an extra β-chain gene, which pairs with a DR-α chain demonstrating that the three -DR, -DP, and -DQ genes can actually give rise to four HLA class II molecules. HLA class III antigens are HLA-linked antigens, but are related to the complement system and less relevant to transplantation. Antigens encoded by non-MHC loci, called minor histocompatibility antigens, elicit a weaker immunologic response, but nevertheless can contribute to transplant rejection by the sheer magnitude of minor antigens. Clinically antigenic disparity in the setting of known HLA identity is recognized. This disparity is attributed to the incidence of graft versus host disease (GVHD) in HLA-identical bone marrow transplants, as well as the high incidence (greater than 30%) of rejection episodes in HLA-identical kidney transplants.
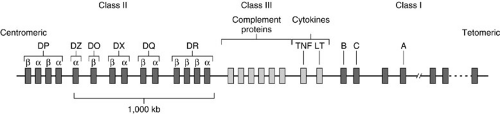 FIGURE 45-2. The major histocompatibility region can be divided into several classes based on functional differences of the proteins produced by the different genes. The human leukocyte antigen class I genes are found at the telomeric end of the short arm of chromosome 6, whereas the class II genes are found at the centromeric end. Also found in this region are the genes for the class III (complement) proteins and some cytokines. TNF, tumor necrosis factor; LT, leukotriene. (Adapted from Abbas AK, Lichtman AH, Pober JS. Cellular and molecular immunology. Philadelphia: WB Saunders, 1991:110.) |
The hallmark of the MHC is its high polymorphism with different class I and II genes. The polygeny (more than 200 alleles) and polymorphism of the MHC region are distinct to this genetic locus; both contribute to the genetic diversity of MHC molecules. The specific combination of MHC alleles on a chromosome is a MHC haplotype.
The antigenic specificity of an MHC haplotype is determined by those genes encoded in the MHC creating an individual fingerprint such that every individual has a unique set of MHC molecules. MHC genes are inherited by classic Mendelian genetics. Expression of MHC alleles is codominant; therefore, each individual inherits three MHC class I genes (-A, -B, -C) and three MHC class II genes (-DR, -DQ, -DP) from each parent. Thus, an individual expresses typically six different MHC class I molecules and from six to eight different class II molecules. Notably, class II gene expression is highly variable with potentially multiple (10,11,12,13,14,15,16,17,18,19,20) class II proteins expressed by one individual.
The antigenic specificity of an MHC haplotype is determined by those genes encoded in the MHC creating an individual fingerprint such that every individual has a unique set of MHC molecules. MHC genes are inherited by classic Mendelian genetics. Expression of MHC alleles is codominant; therefore, each individual inherits three MHC class I genes (-A, -B, -C) and three MHC class II genes (-DR, -DQ, -DP) from each parent. Thus, an individual expresses typically six different MHC class I molecules and from six to eight different class II molecules. Notably, class II gene expression is highly variable with potentially multiple (10,11,12,13,14,15,16,17,18,19,20) class II proteins expressed by one individual.
The protein structure of class I and II glycoproteins is similar and is a heterodimer, a structure typical of many immune receptors. The class I molecular structure contains a MHC-encoded α (heavy) chain noncovalently associated with a β chain, the invariant β2-microglobulin. The β2-microglobulin is located at a different locus in humans and is essential for cell surface expression of class I molecules. The protein structure of the α chain has three extracellular glycosylated domains (α1, α2, α3), a hydrophobic transmembrane region, and a cytoplasmic tail. X-ray crystallography has elucidated the three-dimensional structure of the class I molecule and the peptide antigen-binding groove, which consists of the two extracellular heavy chain domains, α1 and α2, binding a 8-20 amino acid fragment (16,17). Class II molecules consist of two noncovalently linked chains (α and β), which are divided into four extracellular domains (α1, α2 and β1, β2), a hydrophobic transmembrane region, and cytoplasmic tail (18).
The class I and II molecules are structurally similar with four major extracellular domains (class I: α1, α2, α3, and β2 microglobulin and class II; α1, α2, β1, and β2) of which two of the domains (class I: α1, α2 and class II: α1, β1) contribute predominantly to the peptide-binding groove (19,20). The genetic polymorphism is localized primarily to the α1 and α2 domains of class I molecules and the α1 and β1 domains of class II molecules. These domains with the known highest polymorphism form the peptide-binding clefts. Peptides bind to MHC molecules via anchor residues. These anchor residues are sequence motifs and thus are key conserved residues that share structural features essential for function. The peptide-binding groove of MHC class II molecules accommodates more length heterogeneity, such as 12-24 amino acids. The groove is larger and a more open structure, allowing for more permissive binding.
The structural constraints of the peptide-binding groove suggest that certain antigens may be preferentially presented by certain MHC molecules after processing (21). Endogenous proteins (viral products) are degraded into peptides, bind to class I MHC in the endoplasmic reticulum, and are transported to the surface of an APC. Exogenous proteins (bacteria) are endocytosed, degraded in endosomes, and bound to class II molecules to be expressed on the surface of the APC to stimulate CD4+ T cells. In summary, the two classes of MHC molecules transport peptides from different compartments (class I-cytosolic, class II-endocytic) to the cell surface. These molecular processing pathways for peptides provide the intracellular determinants to recognize a peptide as foreign.
IMMUNOBIOLOGY OF THE MHC
The MHC region is the dominant region determining immune responses to allografts. Cellular immunity mediated by T cells first requires the presentation of foreign proteins on APCs. MHC molecules present foreign antigens to the immune system in the context of self-MHC molecules (22). Doherty and Zinkernagel’s Nobel Prize in 1996 was based on the demonstration that antigen recognition by T cells requires the T cell to share self-MHC alleles with the APC, and thus, the T cell is MHC restricted. Class I antigens activate cytotoxic T lymphocytes (CD8+) and induce strong antibody-mediated rejection, detected by cell-mediated lympholysis assays and cross-matching techniques, respectively. CD8+ T cells recognize MHC class I:peptide complexes derived from cytosolic and processed pathogens, and are activated to kill cells displaying such foreign peptides. Class II antigens primarily activate helper T lymphocytes (CD4+) detected by mixed lymphocyte culture reactions. CD4+ T cells recognize MHC class II:peptide complexes from endocytosed proteins and activate immune effector cells, such as B cells or APCs, against foreign antigens with these peptides. The immune response is highly reactive to foreign MHC. In fact, there is a tenfold increase in T-cell proliferative responsiveness if stimulated with foreign MHC or allo-MHC, as opposed to an exogenous antigen. This alloreactivity is the specific recognition of foreign MHC molecules by the host T cells, which bind either directly to the donor MHC-peptide complex (class I or direct allorecognition) or to processed allo-MHC antigens presented on host APCs (class II or indirect allorecognition).
CELLULAR IMMUNE DEFENSES
There are principally four main subsets of the cellular immune system including T cells, B cells, NK cells, and APCs.
T Cells
T cells undergo maturation and selection in the thymus; upon activation to properly presented antigens by an APC, they then constitute the effector arm of cell-mediated immune responses. T cells are principally divided into T-helper (CD4+) and T-cytotoxic (CD8+) cells.
The T-helper cells stimulate and activate other components of the immune system providing help to other T cells, B cells, and macrophages, whereas T-cytotoxic cells directly lyse cells displaying foreign antigens. However, these functional/phenotypic categories, once believed to be completely distinct, actually may functionally overlap, suggestive of an inherent plasticity between subsets. There are also suppressor T lymphocytes, which downregulate immune responses by direct inhibition: CD4+cells, CD8+cells, and non-T cells can function as autoregulatory cells inducing nonresponsiveness (23,24).
The T-helper cells stimulate and activate other components of the immune system providing help to other T cells, B cells, and macrophages, whereas T-cytotoxic cells directly lyse cells displaying foreign antigens. However, these functional/phenotypic categories, once believed to be completely distinct, actually may functionally overlap, suggestive of an inherent plasticity between subsets. There are also suppressor T lymphocytes, which downregulate immune responses by direct inhibition: CD4+cells, CD8+cells, and non-T cells can function as autoregulatory cells inducing nonresponsiveness (23,24).
The T-helper cells are further subdivided into two subsets based on differential cytokine expression (25). T-helper type 1 (TH1) cells elicit proinflammatory cellular immune responses and allograft rejection by secreting IL2 and γ-IFN. T-helper type 2 (TH2) cells augment B-cell differentiation and maturation, and secrete IL-4, -5, -6, and -10. The TH2 cells are involved in humoral immunity and allograft acceptance. The antiinflammatory cytokines of TH2 cells may be one venue for graft survival during organ transplantation. The two subsets form regulatory circuits such that a given immune response is either predominantly TH1 or TH2, proinflammatory or humoral, depending on a particular antigen (Fig. 45-3). Both γ-IFN and IL-12 (the macrophage-derived T-cell–stimulating cytokine) promote TH1 proliferation, whereas γ-IFN downregulates a TH2 response (26). Conversely, IL-4 secreted by TH2 cells, downregulates a TH1 response and γ-IFN release. So the differential cytokine profile directs the predominance of a TH1 or TH2 response. Just as there are subsets of T-helper cells, subsets of cytotoxic T cells have been always purported, similarly substantiated by the differential cytokine expression from antigen-stimulated CD8+ clones.
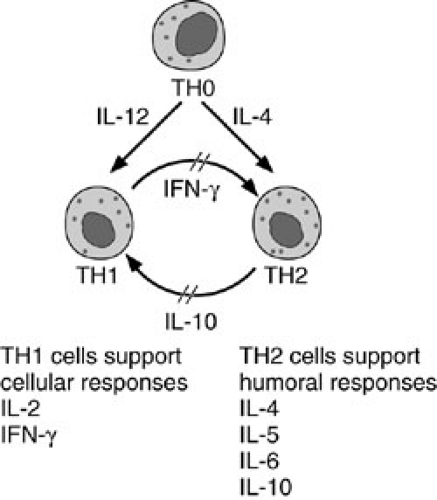 FIGURE 45-3. Differential cytokine production determines T-helper (TH) cell subset differentiation. Interleukin-12 (IL-12), released by macrophages after stimulation by bacteria, protozoa, or viruses, and interferon-γ (IFN-γ), released by natural killer cells and γε T cells, cause the naive (TH0) cell to develop into a TH1 cell. Conversely, helminthic pathogens stimulate γε T cells, basophils, or mast cells and perhaps another, unknown, cell type to release IL-4. This IL drives TH0 cells to differentiate into the TH2 subset. TH2 cells produce IL-10, which inhibits the activation of TH1 cells. TH1 cells produce IFN-γ, which inhibits TH2 activity. |
All T cells express a T-cell receptor (TCR), a glycosylated heterodimer capable of recognition of the MHC-peptide complex. The TCR is a complex of proteins including the antigen-specific receptor (αβ TcR), a CD3 molecule, and associated invariant chains, γ, δ, and ε (27) (Fig. 45-4). There are two types of heterodimeric TCRs: (1) αβ+ (α and β chains), which are the most common; and (2) γδ+ (γ and δ chains). The αβ+ TCR is the antigen-specific receptor on cytolytic and helper cells and represents the majority of T cells in the peripheral blood and lymphoid tissues. The γδ+ TCR is less clear; only a minority of T cells is γδ+. There are γδ+ TCR+ T cells located at areas that interface with the environment, such as gut and bronchial-associated lymphoid tissues.
Most αβ TCR+ T cells mature in the thymus, whereas γδ TCR+ T cells develop extrathymically in the periphery. The pluripotent stem cell produces a precursor T cell, double negative, TCR–/CD4–/CD8–/CD44+, which migrates to the thymus for maturation, rearrangement, and expression of the TCR α and β chains to become double positives TCR+/CD4+/CD8+/CD44–. Negative selection (or clonal deletion) eliminates autoreactive clones strongly reactive with self-MHC by apoptosis and is mediated by bone marrow-derived dendritic cells in the thymic medulla. During positive selection, developing thymocytes interact with host thymic epithelium in the thymic cortex, and those that recognize antigen in context of self-MHC survive and are restricted to self-MHC. Less than 1% of all pre-T cells leave the thymus as functionally mature single positive T lymphocytes, CD4+ or CD8+, with rearranged TCR genes. The diversity of the TCR repertoire is generated by α and β chain rearrangements, as well as by the thymic positive and negative selection mechanisms.
Presentation of an antigen through the MHC complex to T cells confers high specificity, but low affinity. Upon recognition of the antigen-MHC complex, the conformation of the TCR complex changes and intracellular signaling activates the immune response. A secondary costimulatory signal is essential to be provided by an APC before T-cell activation occurs. In its absence, T-cell activation does not occur and the T cells become “anergic” or inactivated (28). This second step of a costimulatory signal is conferred via the CD28 molecule, expressed on mature T cells, to the B7 ligand constitutively expressed on APCs. Only with both signals provided do T cells secrete the optimum concentrations of IL2 essential for proliferation. The CD28 binding to B7 stabilizes the TCR–antigen interactions. All CD4+ cells and one-half of CD8+ cells express CD28; CTLA-4 is another such costimulatory molecule, other than CD28, that binds B7 and is present on activated T cells.
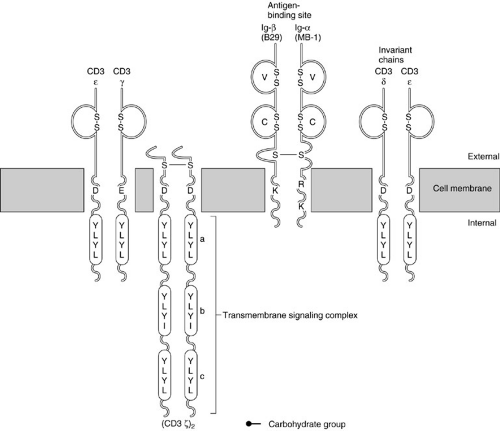 FIGURE 45-4. The T-cell antigen receptor complex comprises four polypeptide dimers (TCR αβ, CD3 γε, CD3 δε, and CD3 ζζ). As shown in this schematic, the antigen-binding site itself is formed from the variable regions of the αβ TCR heterodimer (or the γε TCR heterodimer). The associated CD3 protein is an important cytoplasmic signaling component of the TCR complex, and through interaction with intracellular tyrosine kinases, translates the signals generated by antigen binding into functional changes in cellular activity. C, constant region; V, variable region. (Adapted from Malissen B, Schmitt-Verhulst AM. Transmembrane signalling through the T-cell receptor–CD3 complex. Curr Opin Immunol 1993;5: 324.) |
B Cells
B lymphocytes, central to humoral immunity, develop from the pluripotent hematopoietic stem cell into either a mature plasma cell secreting immunoglobulin or a peripheral resting B cell. The primary function of B cells is to produce a specific clonal antibody in response to antigenic stimulation. Postnatally, B-cell development is initiated in the bone marrow. The pro–B-cell stage is the first committed step in B-cell development with germ-line unrearranged immunoglobulin genes. Subsequent heavy chain V(D) J recombination occurs with cytoplasmic expression of the μ (heavy) chain and results in a pre-B cell. The development from the pre–B-cell lineage to the mature B cell requires rearrangement of the B-cell immunoglobulin genes. The VDJ recombination of the germ-line DNA, as well as the hypervariable regions, is the foundation of the extreme diversity of antibodies. Heavy chain rearrangement precedes light chain rearrangement and only one light chain isotype (predominantly 95% κ light chain) will be expressed on a mature B cell. When the μ (heavy) chain associates with rearranged light chains (λ or κ), then immunoglobulin is expressed on the cell surface of immature B cells.
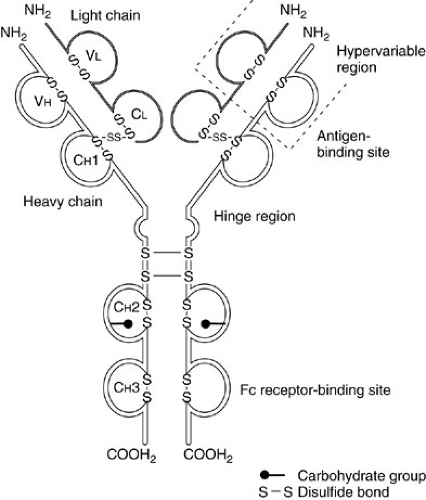 FIGURE 45-5. The immunoglobulin is composed of two heavy and two light chains. The heavy chain constant region determines the isotype of the molecule, whereas the heavy and the light chain variable regions constitute the receptor or antigen-binding portion of the immunoglobulin. CH, heavy chain constant region; CL, light chain constant region; VH, heavy chain variable region; VL, light chain variable region. (Adapted from Abbas AK, Lichtman AH, Pober JS. Cellular and molecular immunology. Philadelphia: WB Saunders, 1991:39.) |
Once the immature B cell actually expresses membrane-bound IgM, it migrates from the bone marrow to the bloodstream. Antigen-dependent B-cell maturation and antigen-independent maturation occur in the periphery. Without antigenic exposure, these cells mature and persist as resting B lymphocytes that are IgM+ or coexpress IgM+IgD+. Stimulation by an antigen is the prerequisite for the final stage of maturation to a plasma cell, secreting immunoglobulin reactive to foreign antigens.
Immunoglobulins are heterodimer glycoproteins composed of two light (L) and two heavy (H) chains, stabilized by several transmembrane glycoproteins. Each chain (L or H) contains variable and constant regions (Fig. 45-5). The variable region with hypervariable areas makes up the antigen-binding site (Fab). The constant region (Fc) determines the isotype and thus class of immunoglobulin, and provides the effector function such as binding complement. There are five immunoglobulin classes, IgM, IgG, IgA, IgE, and IgD, that differ by their heavy chains and by their function. IgM is expressed on resting B cells and is a pentamer. It is the major antibody in a primary antibody response and capable of fixing complement. IgG has multiple subtypes (IgG1, IgG2, IgG3, and IgG4) and is the most prevalent antibody (75%). It is detected in a primary response after isotype switching. IgG is the only immunoglobulin to cross the placenta and can bind as does IgM to phagocytic cells to destroy microorganisms. IgA has two isotypes, IgA1 in the serum and IgA2 (dimer) in secretions. Serum IgA is the second most common antibody (15%). Secretory IgA is secreted locally in mucous membranes and provides a primary barrier for host defense. IgE functions primarily in type I hypersensitivity responses and helminthic destruction. IgD is in minute levels in the serum and expressed on the circulating immature B lymphocytes. For complement activation, the IgG and IgM immunoglobulin can fix complement, thus use the classic pathway, whereas IgA, IgE, and IgD activation use the alternative pathway.
Once the immunoglobulin binds antigen or surface Ig receptors are cross-linked by antigen, then the B-cell receptor complex transduces a signal. B-cell responses may be (1) T-cell–independent, in which B cells alone respond to antigen and are IgM predominant; or (2) T-cell–dependent, requiring B-cell interaction with T cells and APCs (Fig. 45-6).
Signal transduction elicits the transcription of genes essential for proliferation and cellular differentiation into an activated B lymphocyte. Activated B cells undergo isotype switching to differentiate into antibody-producing plasma cells, which constitute a primary antibody response. Antibodies produced in a secondary immune response develop a higher affinity to the antigen, or undergo “affinity maturation.” Somatic hypermutation is the mode of diversification that produces higher-affinity clones.
Natural Killer Cells
NK cells are cytotoxic lymphocytes and phenotypically large granular lymphocytes. They are capable of nonspecifically killing tumors or virally infected cells without prior exposure and without MHC restriction. A subset of NK cells has the ability to destroy allogeneic cells without any reactivity to autologous cells (29). NK cells secrete immunoregulatory cytokines, including γ-IFN, granulocyte-macrophage colony-stimulating factor (GM-CSF), and IL-1, which can directly activate macrophages. Likewise, immunoregulatory cytokines can influence NK activity. NK cells are positively upregulated by IL-12 (NK stimulatory factor) to enhance cytolytic activity and downregulated by IL-10 (a product of TH2 cells), which inhibits γ-IFN secretion.
Antigen-presenting Cells
The prototypic APCs are monocyte/macrophage or dendritic cells that constitutively express both class I and class
II MHC and are capable of phagocytosizing, processing, and presenting antigen fragments as epitopes on their cell surface. Dendritic cells are the most efficient at antigen presentation, even more than macrophages. Virtually any cell type can perform the functions of an APC because antigen processing uses ubiquitous cellular pathways. Even B cells become efficient APCs because surface immunoglobulin can bind low concentrations of antigen with high affinity. Previously known as the reticuloendothelial system, the mononuclear phagocyte system includes macrophages, dendritic cells, hepatic Kupffer cells, tissue histiocytes, and Langerhans cells in the skin. Quiescent endothelial cells in the transplanted graft activated by trauma, ischemia, infection, or any proinflammatory and nonimmunologic event, such as cold ischemia time, become activated for antigen presentation. γ-IFN and TNF-α are immunostimulatory cytokines crucial to macrophage activation. In general, APCs function to phagocytose antigen, process it into 5-15 amino acids fragments, and present those peptides on their cell surface to an effector lymphocyte with a complementary receptor and MHC molecule. APCs also have an antibody-independent NK-like activity with an ability to destroy pathogens directly, as well as the capacity to bind complement and immunoglobulin.
II MHC and are capable of phagocytosizing, processing, and presenting antigen fragments as epitopes on their cell surface. Dendritic cells are the most efficient at antigen presentation, even more than macrophages. Virtually any cell type can perform the functions of an APC because antigen processing uses ubiquitous cellular pathways. Even B cells become efficient APCs because surface immunoglobulin can bind low concentrations of antigen with high affinity. Previously known as the reticuloendothelial system, the mononuclear phagocyte system includes macrophages, dendritic cells, hepatic Kupffer cells, tissue histiocytes, and Langerhans cells in the skin. Quiescent endothelial cells in the transplanted graft activated by trauma, ischemia, infection, or any proinflammatory and nonimmunologic event, such as cold ischemia time, become activated for antigen presentation. γ-IFN and TNF-α are immunostimulatory cytokines crucial to macrophage activation. In general, APCs function to phagocytose antigen, process it into 5-15 amino acids fragments, and present those peptides on their cell surface to an effector lymphocyte with a complementary receptor and MHC molecule. APCs also have an antibody-independent NK-like activity with an ability to destroy pathogens directly, as well as the capacity to bind complement and immunoglobulin.
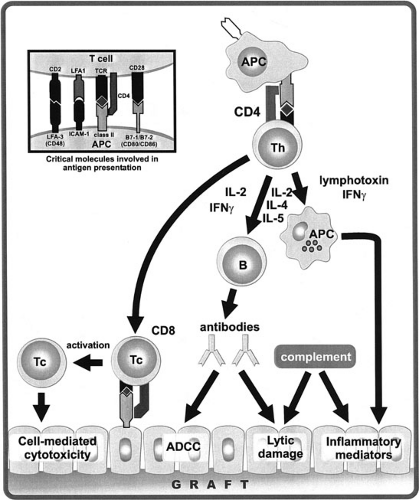 FIGURE 45-6. Association of APCs and T cells involves nonspecific reversible binding through adhesion molecules, such as LFA-1/ICAM and CD2/LFA-3 interactions. Recognition of the peptide antigen-MHC molecular complex by the TCR provides the specificity of the T-cell interaction. Costimulatory molecules (CD28/B7; CTLA-4/B7) mediate a second signal essential for T cells to respond. Then TH cells are activated by APCs in the donor graft to release lymphokines: (1) IL-2 and γIFN to induce cytotoxic T-cell (Tc) activation; (2) IL-2, IL-4, and IL-5 to activate B cells; and (3) γIFN to activate macrophages for nonspecific inflammation. All categories of the immune response reject the graft by cellular, humoral, or nonspecific inflammation. |
EFFECTOR MECHANISMS OF THE IMMUNE RESPONSE
The immune response is a highly integrated and orchestrated series of events. The initial interaction is between the histocompatibility antigens of the graft and the host APCs, such as B cells, dendritic cells, or macrophages (8). Circulating host effector cells may encounter graft antigens either in situ in the transplanted organ or in the draining lymph nodes to which antigens from the graft or passenger lymphocytes drain. Upon antigen exposure, the dendritic cells migrate to the spleen to activate naive T cells and thus amplify sensitization in peripheral
lymphoid organs. Vascular endothelial cells express class II antigens and can function as dendritic cells; their removal has correlated with prolonged vessel allograft survival (30). Likewise, depletion of passenger lymphocytes from the donor alone leads to prolonged survival in islet allografts suggestive of their role in host sensitization. T-cell activation requires both processed antigen presentation by a self-APC and a costimulatory signal, such as a CD28:B7 interaction, stabilizing the antigen–TCR interaction. For allo-MHC antigens, MHC class I antigens recognition is by host CD8+ T cells directly binding to donor MHC-peptide complex. MHC class II recognition occurs when CD4+ T cells bind allo-MHC antigens presented by host APC in context of self-MHC. Alloantigen also binds to surface IgM on B cells, initiating differentiation and maturation into antigen-specific B cells. Thus, the alloantigen binds to the TCR or the surface IgM as the first signal followed by a second costimulatory signal by the APC (31). The recognition of foreign alloantigen results in complex events for both T- and B-cell activation, including the proliferation and differentiation into sensitized antigen-specific CD4+ and CD8+ T cells capable of delayed hypersensitivity reactions and cytolytic activity, respectively. Activation of CD4+T cells requires IL-1 production by the APC. The CD4+T cells initially do not express IL-2 receptors at rest, but upon antigen exposure produce IL-2 and further upregulate IL-2 receptors for clonal expansion. The release of IL-2 initiates the differentiation and activation of CD8+precursors into mature cytolytic T cells. CD4+ T cells also release IL-4 and IL-5 for B-cell activation. Activated B cells proliferate and differentiate into antigen-specific plasma cells. Thus, the differential cytokine secretion patterns of the CD4+ cells as either TH1 cells (IL-2, γIFN) or TH2 cells (IL-4, IL-6, IL-10) directly influence the alloresponse as primarily cellular or humoral, respectively (32).
lymphoid organs. Vascular endothelial cells express class II antigens and can function as dendritic cells; their removal has correlated with prolonged vessel allograft survival (30). Likewise, depletion of passenger lymphocytes from the donor alone leads to prolonged survival in islet allografts suggestive of their role in host sensitization. T-cell activation requires both processed antigen presentation by a self-APC and a costimulatory signal, such as a CD28:B7 interaction, stabilizing the antigen–TCR interaction. For allo-MHC antigens, MHC class I antigens recognition is by host CD8+ T cells directly binding to donor MHC-peptide complex. MHC class II recognition occurs when CD4+ T cells bind allo-MHC antigens presented by host APC in context of self-MHC. Alloantigen also binds to surface IgM on B cells, initiating differentiation and maturation into antigen-specific B cells. Thus, the alloantigen binds to the TCR or the surface IgM as the first signal followed by a second costimulatory signal by the APC (31). The recognition of foreign alloantigen results in complex events for both T- and B-cell activation, including the proliferation and differentiation into sensitized antigen-specific CD4+ and CD8+ T cells capable of delayed hypersensitivity reactions and cytolytic activity, respectively. Activation of CD4+T cells requires IL-1 production by the APC. The CD4+T cells initially do not express IL-2 receptors at rest, but upon antigen exposure produce IL-2 and further upregulate IL-2 receptors for clonal expansion. The release of IL-2 initiates the differentiation and activation of CD8+precursors into mature cytolytic T cells. CD4+ T cells also release IL-4 and IL-5 for B-cell activation. Activated B cells proliferate and differentiate into antigen-specific plasma cells. Thus, the differential cytokine secretion patterns of the CD4+ cells as either TH1 cells (IL-2, γIFN) or TH2 cells (IL-4, IL-6, IL-10) directly influence the alloresponse as primarily cellular or humoral, respectively (32).
Alloantigen recognition by already sensitized cells or antigen-specific memory cells then induces the efferent limb of the immune response (22). As previously stated, allosensitization occurs primarily in the draining lymph nodes, then sensitized lymphocytes enter the recirculating pool to migrate to the graft during the immune response. Upon recognition of the foreign alloantigen, the effector lymphocytes release lymphokines to further recruit both nonspecific inflammatory cells and sensitized lymphocytes in one microenvironment to augment the proinflammatory state. B cells secrete antibody specifically to bind to the allograft and mediate destruction by complement-mediated lysis. An antibody-coated allograft directs other cells (NK, APCs, granulocytes) with Fc receptors to kill by antibody-dependent cellular cytotoxicity. APCs activated by TNF cause local tissue destruction. The combination of both CD4+ and CD8+ allosensitized T cells are the primary effectors of allograft rejection. The relative roles of each T-cell subset in allograft rejection may be determined by the dependence of CD8+ cells on the CD4+ subset for amplification. CD4+ cells mediate allograft rejection through lymphokine release and recruitment of nonspecific inflammatory cells for a delayed-type hypersensitivity response. The role of CD4+ cells (the TH1/TH2 responses) in the alloresponse has expanded our appreciation of the role of cytokines in rejection (32). CD8+ cells effect allograft rejection through direct allograft contact by release of cytotoxic granules or engaging the Fas receptor to induce apoptosis (33).
CLINICAL ALLOGRAFT REJECTION
The diversity of immune effector mechanisms results in three fundamental presentations believed to differ in timing and underlying conceptual immune mechanisms. Hyperacute rejection occurs instantly upon reestablishing vascular flow to the transplant. The organ becomes grossly cyanotic, swollen, and soft, with ensuing vascular thrombosis. Preformed antibodies present in the host’s serum bind to the allograft instantly and activate the complement cascade with immediate graft destruction. Hyperacute rejection occurs after transplantation of ABO-incompatible grafts or if the recipient possesses high titers of antidonor class I HLA antibodies (34). Anti-HLA antibodies do not occur naturally without prior exposure to foreign histocompatibility antigens. Preformed antidonor antibodies are found typically in patients with a history of multiple transfusions, prior organ rejection, and multiparity because these women are exposed to in utero foreign antigens. Such antibodies in the recipient, reactive to donor antigens, are detected by a positive cross-match, which indicates the presence of specific sensitization to donor histocompatibility antigens. A positive cross-match can be detected by a lysis of donor lymphocytes occurring in the presence of recipient serum (via complement-dependent lymphocytotoxic assay). Plasmapheresis is a current technique to remove preformed antibodies in ABO-incompatible transplants (35). Liver transplants, unlike kidney, heart, lung, and pancreas, are relatively unique in their resistance to hyperacute rejection and can occur despite ABO incompatibility or a positive cross-match. Hyperacute rejection causes immediate graft destruction for which there is no rescue therapy. It represents the formidable immunologic barrier in the experimental application of xenotransplantation (36). Most mammals naturally have high titers of preformed antibodies directed against cell surface antigens of other discordant species. Both the naturally species-specific preformed antibodies and resultant activation of the complement cascade need to be circumvented prior to successful xenotransplantation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


