Necrotizing Enterocolitis
Shawn J. Rangel
R. Lawrence Moss
Section of Pediatric Surgery, Yale University School of Medicine, New Haven, Connecticut 06520.
Section of Pediatric Surgery, Yale University School of Medicine, New Haven, Connecticut 06520.
In the mid-1970s, necrotizing enterocolitis (NEC) emerged as the most common surgical emergency in the neonatal intensive care unit (NICU) (1,2). Almost three decades later, and despite a growing knowledge base supported by hundreds of laboratory and clinical research studies, NEC continues to pose a significant challenge to the pediatric surgeon with respect to its clinical management. At present, NEC remains the major cause of death for all neonates undergoing surgery, and the mortality from this disease is greater than from all congenital anomalies of the gastrointestinal (GI) tract combined (3).
NEC is almost exclusively a disease of prematurity. Little was known or documented about the disease in the first half of the twentieth century because survival was unlikely in the extremely premature. Subsequent advances, including development of modern neonatal respiratory care, allowed for the survival of babies of ever-decreasing gestational age and birth weight. A striking increase in the incidence of NEC was then observed, as these breakthroughs led to an increasing number of infants at risk for the disease.
The strength of epidemiologic evidence implicating prematurity as a risk factor for NEC is substantial. Premature infants comprise greater than 90% of all cases, and nearly 90% of these have birth weights less than 2,000 g (2,4). Prematurity is the only risk factor consistently identified in case-control studies, and the disease is relatively rare in countries where prematurity is uncommon (Japan and Sweden) (5). The incidence of NEC is estimated between 1% to 8% percent of NICU admissions (1 to 3 in 1,000 live births) (6,7). Incidence is higher in previously fed babies (15%) and in those weighing less than 1,500 g (10% to 15%) (8,9). NEC most commonly affects babies born between 30 and 32 weeks’ gestation, and is diagnosed most often during the second week of life (10,11). Epidemiologic studies have characterized an inverse relationship between the degree of prematurity and the age of onset of NEC, with more premature infants developing NEC at relatively later postnatal ages (12).
The financial burden attributable to this disease is substantial. It has been estimated that the additional cost of treating NEC in a premature neonate ranges from $74,000 to $186,000 compared with age-matched controls without the disease (13). With current estimates of incidence rates, this translates into roughly one-third of 1 billion per year in health care-related costs in the United States. The overall financial impact of this disease is likely to be much greater, however, because this estimate does not take into account the cost of caring for infants with lifelong morbidity from NEC.
ANATOMY AND HISTOLOGY
NEC can affect any segment of the GI tract, from the stomach to the distal rectum. Injury most commonly involves both the small and large bowel together (40% to 45% of cases), followed by isolated lesions of the small intestine (30%) and colon (25%) (14). The ileocecal region is the most commonly affected area and is often the site of the most severe injury. Approximately 35% of patients will have focal disease at laparotomy, and a similar proportion will have multisegment disease localized to a relatively small length of bowel (14,15). In 15% to 20% of patients, exploration will reveal massive necrosis involving a considerable portion of the entire bowel. Such paninvolvement (NEC totalis) is often fatal.
Coagulation necrosis is the hallmark histologic finding in NEC and is present in more than 90% of pathology specimens (14). Coagulation necrosis is invariably associated with ischemic injury, suggesting the importance of diminished splanchnic perfusion in the pathogenesis of NEC. Bacterial overgrowth is found in approximately two-thirds of specimens. The depth of injury to the bowel can be variable, and areas of full- and partial-thickness necrosis
may be found adjacent to frank perforation. In contrast to other causes of intestinal necrosis (infectious agents and ischemic necrosis), there is an exaggerated inflammatory response that extends to grossly normal areas of bowel. Pneumatosis intestinalis is found in approximately one-half of specimens and is considered pathognomonic for NEC. Mixed areas of acute and chronic inflammatory changes are common, and epithelial regeneration is found in more than one-half of specimens (14). These findings suggest the pathogenesis of NEC may involve a chronic process of injury and repair rather than a single critical insult, in contrast to a thromboembolic event.
may be found adjacent to frank perforation. In contrast to other causes of intestinal necrosis (infectious agents and ischemic necrosis), there is an exaggerated inflammatory response that extends to grossly normal areas of bowel. Pneumatosis intestinalis is found in approximately one-half of specimens and is considered pathognomonic for NEC. Mixed areas of acute and chronic inflammatory changes are common, and epithelial regeneration is found in more than one-half of specimens (14). These findings suggest the pathogenesis of NEC may involve a chronic process of injury and repair rather than a single critical insult, in contrast to a thromboembolic event.
PATHOGENESIS
Despite extensive clinical and laboratory investigation, a complete understanding of the pathogenesis of NEC remains elusive. The etiology of NEC has classically been attributed to a maladaptive response of the premature gut to the presence of feeding substrate and bacterial colonization. Although these risk factors are almost invariably present in documented cases of NEC, the overwhelming majority of premature babies who are exposed to these factors do not develop the disease. More contemporary models of pathogenesis have therefore placed greater emphasis on a multifactorial etiology, stressing the role of ischemia, reperfusion injury, and the complex cascade of inflammatory mediators. A synopsis of the laboratory and clinical evidence supporting current theories of pathogenesis is presented in the next section.
Ischemia and Reperfusion Injury
The role of ischemia and reperfusion injury in the pathogenesis of NEC has largely been characterized through neonatal animal models. The molecular events leading to cellular damage have been well characterized, although the factors initiating this process in the context of clinical NEC are still unknown. Hypoxia at the cellular level leads to a marked increase in the production of xanthine oxidase (XO). Upon reperfusion, XO converts hypoxanthine to xanthine and uric acid, releasing a host of reactive oxygen species in the process (hydroxyl, superoxide, and hydrogen peroxide radicals). These species can induce substantial damage to lipoprotein components of the cell membrane, increasing permeability and facilitating bacteria translocation. Even relatively brief periods of hypoxia can lead to prolonged and substantial changes in mucosal permeability through this mechanism (16).
The precise mechanism and temporal relationship for ischemic injury in the context of NEC remains unclear. Gross and histopathologic findings do not support a thromboembolic process or a generalized low-flow state as a primary mechanism of injury. In this regard, mesenteric infarction and low-output states (previously believed to be associated with NEC through umbilical artery catheter use and cardiogenic shock, respectively) appear to be distinct clinicopathologic entities. Increasing evidence suggests that ischemic injury associated with NEC may result from a maladaptive vascular response to early pathogenic events in the disease. As such, ischemia may play a role in the progression of NEC rather than serving as a critical precipitating factor.
Several theories have been proposed to explain the etiology of ischemia associated with NEC and prematurity. Hyperactive extrinsic vascular regulation (the so-called “diving” or “Herring-Breur” reflex) has been implicated because of its potential relevance to the neonate (17,18). Initially described in diving mammals, the reflex involves a preferential diversion of blood flow to the heart and brain during periods of severe hypoxia. This presumably occurs during the period of parturition. As such, this theory cannot adequately explain the clinical observation that NEC commonly occurs during the second week of life. Furthermore, significant episodes of hypoxia prior to bowel injury cannot be identified in most cases of NEC.
Other studies have attempted to characterize the vasoregulatory responses of the premature gut to hypoxia and prolonged sympathoadrenergic stimulation. Studies using neonatal pigs have identified a maladaptive response to these insults in the way of prolonged and exaggerated vasoconstriction (19). Such responses may exacerbate transient (and likely physiologic) episodes of hypoxia. Potential mechanisms include the presence of labile myogenic vascular reflexes, which may be particularly sensitive to changes in venous pressure (20). Such maladaptive vasoregulatory reflexes are not observed in the intestine of mature swine. Protection appears to be conferred through autoregulatory “escape” mechanisms, which are hardwired into the mature intestine (19). Future studies will need to further characterize how complex vasoregulatory mechanisms regulate the intestinal microcirculation, and how these may become dysfunctional in the premature infant.
Inflammatory Mediators
Several inflammatory mediators are elevated in the serum of premature neonates with NEC. These include tumor necrosis factor-alpha (TNF-α), platelet-activating factor (PAF), and the interleukins 6 and 8 (IL-6 and IL-8), among others (21,22,23). Intestinal cells of premature infants appear to elaborate higher concentrations of proinflammatory cytokines (particularly IL-8) in response to endotoxin and IL-1 compared with mature cells (21). The primary challenge has been to distinguish between those that play a pivotal role in the development and progression of NEC, and those that are nonspecific markers of inflammation. This is particularly challenging given the complexity and
redundancy of the inflammatory cascade. Furthermore, many conditions associated with NEC can increase serum levels of inflammatory mediators through independent mechanisms (e.g., sepsis).
redundancy of the inflammatory cascade. Furthermore, many conditions associated with NEC can increase serum levels of inflammatory mediators through independent mechanisms (e.g., sepsis).
Data from animal models have identified PAF as a leading candidate for initiating the early pathogenic events in NEC. PAF is a potent vasoconstrictor of the mesenteric circulation and increases mucosal permeability (24). Infusion of PAF into the intestines of neonatal pigs leads to injury resembling NEC, and will also exacerbate the severity of injury in response to ischemia-reperfusion challenge (25,26). Pretreatment with PAF antagonists (WEB-2086) significantly attenuates injury in both models (27,28). These observations suggest that PAF may be integral to the pathogenic changes seen with ischemia-reperfusion injury. PAF may also influence the progression of NEC through secondary inflammatory pathways (particularly TNF-α) and as a potent chemokine for neutrophil activation (25,26). The propensity of NEC to involve the distal small bowel may reflect the relatively high concentration of PAF receptors in this area (29).
PAF-acetylhydrolase is the enzyme responsible for the metabolism of PAF, and serum levels of this enzyme are markedly attenuated in prematurity (30). Levels are even lower in premature neonates diagnosed with NEC. PAF-induced injury may therefore be due to an increase in functional activity rather than overproduction. The deleterious effects of PAF may be dependent on the presence of other factors, including bacterial endotoxin (31,32). Intestinal injury is not observed when PAF is given to germ-free rats (32). Furthermore, injury to mucosal cells in the presence of bacterial endotoxin may likewise be dependent on the presence of PAF. Pretreatment with PAF antibodies significantly reduces injury following the infusion of endotoxin (lipopolysaccharide) into the guts of neonatal pigs (33).
Nitric oxide (NO) has received much attention for its apparent protective function during ischemic stress. Endogenous NO is produced by three different isoforms of the enzyme nitric oxide synthase (NOS). Sources include endothelial cells, phagocytic cells in the circulation and tissues, and others. The protective effects of NO are mediated through vascular smooth muscle relaxation. This property may be important for counteracting the influence of vasoconstricting cytokines during periods of inflammation and ischemia. NO may also protect mucosal cells by directly modulating PAF activity and limiting neutrophil adhesion to the vascular endothelium (34).
Evidence for the protective role of NO is found in several observations from animal models: (1) The severity of mucosal injury is inversely related to the activity of NOS in tissue preparations; (2) inhibitors of NOS such as L-arginine significantly worsen PAF-induced intestinal necrosis; and (3) NO donors significantly reduce PAF-induced intestinal necrosis (35,36,37). These observations led to the postulate that the NOS regulatory pathway may be dysfunctional in premature neonates. However, the activity of NOS was found to be increased in premature neonates with documented NEC (38,39). It is possible that this observed increase could be inappropriately low when compared with a similar response in mature animals. However, more recent laboratory studies have suggested that the role of NO in the context of NEC may not always be protective. Higher activity levels of the inducible form of NOS have been correlated with increased severity of injury in the neonatal rat model (39). The reaction between NO and reactive oxygen species to generate even more reactive peroxynitrate compounds has been postulated as a potential mechanism for injury. These observations suggest that, much like molecular oxygen, the physiologic effects of NO may depend on the local cellular milieu. Further studies need to clarify the precise role of NO in the context of NEC, and how this protective mechanism may be exploited for potential therapeutic strategies.
Infectious Agents
Bacteria appear to be critical in the pathogenesis of NEC, given the observation that the disease does not occur before intestinal colonization is established. The degree of bacterial overgrowth in NEC appears to exceed that observed with other diseases associated with intestinal necrosis (14). Furthermore, the pathologic finding of pneumatosis in association with intestinal necrosis is exceedingly rare, except in the setting of NEC. Pneumatosis results from the accumulation of hydrogen gas in the intestinal wall due to fermentation of carbohydrates by intestinal flora. With stasis and significant bacterial loads, intraluminal pressure may increase to the point of impeding venous return, thereby promoting ischemia. However, the ability of colonizing bacteria to ferment lactose has not been correlated with the development of NEC. Furthermore, evidence for bacterial overgrowth is absent in up to one-third of pathology specimens (14).
These observations suggested that other bacterial factors may be important for initiating the pathogenic changes in NEC. Certain strains of Escherichia coli and Clostridia can cause intestinal necrosis directly by the elaboration of potent exotoxins (40). These organisms have been identified in relative “epidemics” of NEC affecting NICUs (41). Such epidemics have been halted with the implementation of organism-specific antibiotics and the appropriate hygiene measures. However, cultures obtained from blood, stool, and peritoneal aspirates in most cases of spontaneous NEC do not reveal pathogenic organisms. The most commonly isolated species include E. coli, Klebsiella, Enterobacter, Pseudomonas, Clostridium, and coagulase-negative Staphylococci, among others (42,43). This would suggest two different mechanisms of injury with a shared final common pathway. It is likely that, in true cases of NEC, normally nonpathogenic bacteria become
functionally pathogenic due to changes in the mucosal barrier that occur early in the course of disease.
functionally pathogenic due to changes in the mucosal barrier that occur early in the course of disease.
Physiology of the Premature Gut
Several physiologic deficiencies have been characterized in the gut of premature neonates (Fig. 79-1). Peristalsis and other aspects of intestinal motility can be substantially compromised, particularly when neonates are born prior to their eighth month of gestation (44). Clearance of bacteria may be impeded, potentially leading to stasis and bacterial overgrowth. Gut-associated immunity may also be affected. Relatively fewer functional B lymphocytes are present in the immature gut, and the ability to produce sufficient amounts of secretory IgA is reduced (45). This antibody is believed to protect mucosal cells by preventing the binding of pathogenic organisms. The production of pepsin, gastric acid, and mucus are also decreased in prematurity (46,47). These factors may have important secondary roles in limiting the proliferation of intestinal flora and their ability to bind to mucosal cells. Intestinal trefoil factor is a peptide that has recently been characterized in the mature rat intestine (48,49). The peptide appears to have several adaptive functions, including protecting the mucosal barrier from microbial invasion and limiting the production of reactive oxygen species during ischemic stress. Significantly lower levels of mRNA for this peptide have been found in the prenatal rat. In concert with potentially dysfunctional vasoregulatory mechanisms, these characteristics may collectively act to lower the threshold for NEC in premature infants.
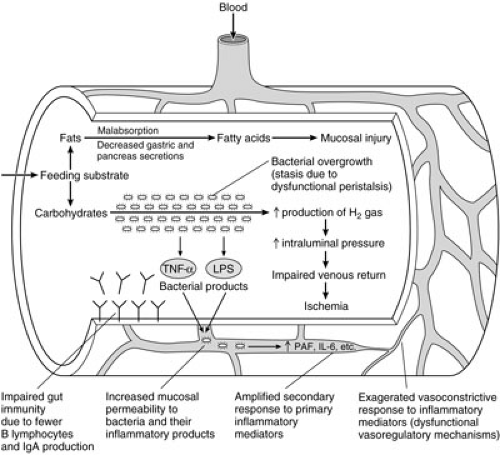 FIGURE 79-1. Characteristics of the premature gut that may increase the risk for developing necrotizing enterocolitis. TNF-α, tumor necrosis factor-alpha; LPS, lipopolysacharide; PAF, platelet-activating factor; IL-6, interleukin 6. |
Role of Feeding
Epidemiologic studies have identified feeding as an important factor in the development of NEC. The majority of neonates who develop this disease have been previously fed, and the relative incidence of NEC is as much as 50% higher in this cohort (9). The relationship between feeding and NEC is complex, and appears to be dependent on the both the composition and quantity of substrate. Infusion of highly concentrated synthetic formulas leads to spontaneous endotoxemia and increased levels of PAF in healthy preterm infants (50,51). Other studies have demonstrated that high-osmolarity formulas can directly injure the villus border of mucosal cells (46). Malabsorption is common in premature neonates, and high concentrations of undigested fatty acids may also contribute to mucosal injury. Increased production of hydrogen gas from the fermentation of large carbohydrate loads may occur in the presence of stasis and bacterial overgrowth. Multiple clinical series have sought to determine whether the rate with which feedings are advanced in premature infants relates to the occurrence of NEC. Results have been inconclusive. Refer to the “Prevention” section later in this chapter.
Pharmacologic Agents and Other Risk Factors
Clinical and laboratory studies have identified a multitude of pharmacologic agents as possible risk factors for NEC. Case-control studies have shown an increased risk for NEC in neonates exposed to indomethacin both postnatally and prenatally (maternal tocolysis) (46,52,53). Indomethacin blocks prostaglandin synthetase and has been shown to impair mesenteric blood flow by increasing mesenteric vascular resistance (54). However, an association suggesting causality in the development of NEC has not held up in more rigorous clinical studies (55).
Other pharmacologic agents have been proposed to cause NEC through a variety of mechanisms. Methylxanthine compounds increase bacterial loads by altering gut motility and can damage enterocytes directly by their intraluminal metabolites (56). Vitamin E has been shown to interfere with intracellular bacterial killing in phagocytic cells (57). The hyperosmolarity of some drug preparations has been postulated to injure the villus border of mucosal cells (58). This potential mechanism is less plausible, however, given the relatively small volumes associated with clinical doses for most agents. Vasopressors and other agents used for hemodynamic support may exacerbate intestinal injury through their effects on the mesenteric circulation. Despite these potential mechanisms, no good clinical evidence exists to suggest that any of these agents play an important role in the initiation of NEC.
Case-control studies have identified relatively weak and inconsistent associations for other potential risk factors. These include hypoglycemia, premature rupture of membranes, chorioamnitis, hypercoagulable conditions, low-output states, prenatal cocaine exposure, and the use of umbilical catheters, among others (59,60,61,62,63,64). The intestinal injury associated with many of these “risk factors” is likely mediated through thromboembolic or vasoconstrictive mechanisms. Although clinically apparent injury may occur through the same final common pathway (ischemia and reperfusion), the initial pathogenic events for what we define as NEC may be quite different. This may suggest the misclassification of disease (and associated risk factors) in many earlier observational studies. Furthermore, the interpretation of existing clinical studies can be challenging for a number of other reasons. These include the frequent use of retrospective study designs, the frequency and severity of comorbid conditions, and difficulty with modeling the ever-increasing complexity of the neonate–NICU interaction, among others.
Unifying Concept of Pathogenesis
Our understanding of the pathophysiology of NEC is improving at a rapid pace. The experimental and clinical evidence outlined previously has led to a better understanding of the events that may be important for the initiation and progression of disease. Although the precise nature and temporal relationship of these events are yet to be determined, a unifying concept of pathogenesis is emerging from currently available data.
The initial insult leading to NEC may be a subclinical event such as a brief episode of perinatal hypoxia or postnatal infection. The injury from this event may not be clinically relevant in neonates with normally developed intestines. Upon colonization of the gut, bacteria may bind to injured mucosal cells and elicit a localized inflammatory response in reaction to endotoxin and other bacterial products. This may lead to further inflammation as the affected endothelial cells release proinflammatory cytokines, including TNF-α and PAF. In the presence of abnormal counterregulatory mechanisms (e.g., reduced PAF-acetylhydrolase), these factors may increase the permeability of mucosal cells leading to the translocation of bacteria and bacterial products. The inflammatory response is then further amplified, leading to the recruitment and activation of circulating neutrophils. Activation of neutrophils may further injure damaged mucosa through the release of secondary mediators and reactive oxygen species. A maladaptive vasoconstrictive response may then follow, further exacerbating injury through ischemia and reperfusion. The end result is a vicious cycle of positive feedback mechanisms that can ultimately lead to frank necrosis and perforation. This process may evolve over days, with injury and repair maintained in a fine balance. Other factors that may compromise mesenteric perfusion (sepsis) or increase oxygen demand (feeding) may tip the balance toward progressive and irreversible injury.
CLINICAL DIAGNOSIS
The diagnosis of NEC is dependent on the signs and symptoms characteristic of intestinal ischemia in the neonate. These may include gross or occult GI bleeding, high gastric residuals, feeding intolerance, diarrhea, and abdominal distension, among others. These signs have little predictive value for NEC as isolated clinical findings. However, NEC should be seriously considered when any of these manifest in a premature neonate with signs of evolving and unexplained sepsis. Specific to the physical exam, the finding of a fixed abdominal mass and erythema of the abdominal wall are the most predictive of NEC when present (near 100% specificity) (65). The diagnostic utility of these findings are limited by their poor sensitivities, however, being absent in as many as 90% of patients with documented disease. A sudden, unexplained need for increased ventilatory support in the premature neonate may also serve as a harbinger of NEC (66).
Symptoms can take on a fulminate course or remain remarkably insidious during this period. The neonate’s inability to mount an effective inflammatory response and to develop physical signs of peritonitis may further complicate the clinical picture. These observations may make it difficult to differentiate NEC from other sources of neonatal sepsis. To aid in clinical diagnosis, Bell and colleagues devised a clinical staging system based on physical exam findings, laboratory data, and radiographic evidence of NEC (Table 79-1). Developed in 1978, the scale remains in wide use today for predicting the likelihood and severity of NEC (67).
Differential Diagnosis
NEC must be differentiated from other conditions associated with abdominal distention and sepsis. The most common challenge faced by the clinician is differentiating NEC from sepsis of a different etiology associated with ileus. Early NEC and sepsis with ileus can appear clinically indistinguishable. In many cases, differentiation between the two is only possible after observing the course of the disease in each patient.
The term focal intestinal perforation (FIP) has been applied to the surgical finding of a single isolated perforation occurring in a premature infant that is not associated with the radiographic finding of pneumatosis. Much debate has ensued as to whether FIP is simply a “mild” version of NEC or whether it is a distinct disease entity. Clinical studies have observed that perforations associated with FIP occur relatively earlier than NEC, and the lesions occur almost
invariably on the antimesenteric side of the distal ileum (68,69). Some reports have also described a different histologic profile associated with FIP, with coagulation necrosis and pneumatosis being notably absent in most specimens (68,69). The histology of NEC is very nonspecific, however, and early lesions may not exhibit the classic findings of well-established NEC (following secondary ischemic injury). Patients with FIP clearly have a better prognosis than those with NEC, although this may simply reflect the difference in the severity of bowel necrosis in the two cohorts. Ultimately, there may be little clinical relevance in distinguishing these processes, as the principles of management are the same for both.
invariably on the antimesenteric side of the distal ileum (68,69). Some reports have also described a different histologic profile associated with FIP, with coagulation necrosis and pneumatosis being notably absent in most specimens (68,69). The histology of NEC is very nonspecific, however, and early lesions may not exhibit the classic findings of well-established NEC (following secondary ischemic injury). Patients with FIP clearly have a better prognosis than those with NEC, although this may simply reflect the difference in the severity of bowel necrosis in the two cohorts. Ultimately, there may be little clinical relevance in distinguishing these processes, as the principles of management are the same for both.
TABLE 79-1 Clinical Staging System for Necrotizing Enterocolitis. | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
Laboratory Studies
Laboratory parameters in patients with NEC are often nonspecific and generally indicate the presence of an inflammatory condition. Leukocyte and platelet counts may be elevated, normal, or low, depending on the severity of NEC and associated sepsis. Leukocytosis is the most common abnormality in NEC and is frequently accompanied by a refractory metabolic acidosis (65). Anemia may also be present if there is significant hemorrhage from the bowel wall. Severely depressed leukocyte and platelet counts have been associated with advanced disease and a worse overall prognosis (70,71). However, none of these individual parameters have sufficient sensitivity, specificity, or predictive accuracy to be of useful diagnostic value.
More recent efforts have attempted to identify novel serum markers for the earlier diagnosis and treatment of NEC. Neonates with confirmed stage II and III NEC were found to have significantly elevated serum levels of PAF compared with age-matched controls without the disease (72). However, only 3 of 11 (27%) patients in this case-control study had elevated levels prior to the onset of clinical signs. Furthermore, the lack of data regarding PAF levels in other causes of abdominal sepsis brings into question the specificity of this assay for NEC. Serum fatty acid-binding proteins have also been examined as a potential diagnostic marker for NEC. Similar to the PAF assay, levels were elevated in only a small proportion (12.5%) of patients with stage I disease (73). Further study is required to better define the diagnostic utility of these assays.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


