Transient Tachypnea of the Newborn
INTRODUCTION
Transient tachypnea of the newborn (TTN) was the name given by Mary Ellen Avery in 1966 to describe a similar clinical presentation in a group of 8 neonates with: marked tachypnea on the first day of life (80–140 breaths/minute), mild cyanosis, mild work of breathing, no evidence of infection, similar chest x-ray findings, and resolution by 2 to 5 days.1 It was also known as type II respiratory distress in the early years in an effort to differentiate it from the better-known respiratory distress syndrome (RDS). It is the most common cause of respiratory distress in the term infant and a frequent reason for admission to the neonatal intensive care unit (NICU).2,3 TTN results from a failure of the normal transition from placental gas exchange in utero to pulmonary gas exchange and breathing. The primary mechanism causing TTN is delayed resorption of fetal lung fluid, a complex process that is now understood to begin several days before spontaneous delivery.4,5 Although our understanding of the processes involved in the clearance of fetal lung fluid has increased, the clinical picture has remained much the same as described in 1966 (Table 24-1).
Table 24-1 Key Diagnostic Features of Transient Tachypnea of the Newborn
Tachypnea with respiratory rate > 60 breaths/minute, often > 100 breaths/minute
Typical chest x-ray findings (hyperinflation, increased perihilar markings, diffuse mild opacities, residual pleural fluid in the interlobar fissures)
Hypoxemia and increased work of breathing (usually mild)
No evidence of systemic illness or infection
Onset in the first few hours of life and lasting more than 6 hours
Complete resolution usually by 72 hours, always by 7 days
EPIDEMIOLOGY
Incidence
Transient tachypnea of the newborn is the most common cause of respiratory distress in the full-term neonate (37–41 weeks) and is responsible for almost half (42.7%–50.3%) of NICU admissions for respiratory distress at term, with an overall incidence of 4.3 to 5.7/1000 live births.6,7 The majority of affected infants are admitted to NICUs for 48 to 72 hours and receive therapies that include respiratory support, intravenous fluids, and antibiotics, resulting in a substantial health care burden. Incidence in premature infants and those with risk factors is substantially higher but often underreported because of overlap with other respiratory disorders associated with prematurity.
Multiple studies have shown that the risk of all types of respiratory distress, and of TTN specifically, increases with each week less than 39 weeks’ gestation.6,8,9 The Consortium on Safe Labor evaluated respiratory outcomes at delivery in 233,844 infants with gestational age greater than 34 weeks, with a special focus on late-preterm infants (34 to 36 and 6/7 weeks) and found the incidence of TTN was highest at 34 weeks (64/1000 births) and 35 weeks (46/1000 births) and decreased with advancing gestational age. The gestational age effect is augmented when delivery is by elective cesarean section, so that even infants delivered at 39 weeks (39 and 0/7 to 39 and 6/7 weeks) have almost double the risk of TTN than their vaginally delivered counterparts.9 TTN in more premature infants, those under 34 weeks, is likely significantly underdiagnosed, as the clinical picture is often complicated by surfactant deficiency and RDS. However, retained fetal lung fluid may contribute significantly to respiratory illness in these infants.
Risk Factors
A number of maternal and perinatal factors have repeatedly been associated with an increased risk of the development of TTN (Table 24-2).10–12 The majority of risk factors are related, directly or indirectly, to relative fetal immaturity and lack of preparedness for labor. Modifiable risk factors, such as mode of delivery, glycemic control in a diabetic mother, and in some cases, prematurity, deserve special attention.
Table 24-2 Risk Factors for Transient Tachypnea of the Newborn
Prematurity
Maternal diabetes
Maternal asthma
Macrosomia
Polyhydramnios
Cesarean delivery
Male gender
Low Apgar score
Cesarean Section
Elective cesarean section performed before 39 weeks is one of the clearly modifiable risk factors in the development of TTN; for this reason, the current American Congress of Obstetricians and Gynecologists (ACOG) guidelines specify such deliveries should occur after 39 completed weeks.13 However, the relationship between cesarean delivery of all types and TTN is complex. It is clear that cesarean delivery increases the risk of respiratory distress at birth, which is especially pronounced at lower gestational ages.9,14 Multiple works have shown an increased rate of TTN among infants delivered by cesarean as well as an increased rate of RDS.15 Late-preterm infants (34–36 and 6/7 weeks) and early term infants (37–38 and 6/7 weeks) are at increased risk of TTN regardless of delivery type, but especially after elective repeat cesarean.8,16 However, it is unclear whether onset of labor prior to cesarean delivery is protective against developing TTN, with some work showing a difference between studied infants but others finding no difference.7,14,15,17
Genetics
Epidemiologic studies provide evidence for a genetic contribution to the risk of developing TTN.18 Familial clustering is seen of cases of TTN without known risk factors,19 and infants born to mothers with a history of asthma have a higher rate of development of TTN.12 Perhaps unsurprisingly, some work showed affected infants are also at a higher risk of developing asthma in childhood.20 Polymorphisms have been studied in a number of genes that produce substances known to have a role in neonatal transition, including surfactant, epithelial sodium channels (ENaCs), and catecholamine.21,22
The majority of studies failed to find an association between polymorphisms of selected introns and TTN. An association was seen for the β-adrenergic receptor (ADRB)21: Infants diagnosed with TTN were significantly more likely to carry loss-of-function polymorphisms for this gene. The role of genetics in TTN, much like the role of genetics in asthma, is complex and multifactorial, with many areas of active research.
PATHOPHYSIOLOGY
The fetal lung is a secretory organ during fetal life, a process essential to normal development of the pulmonary system.23 This must change, and fluid must be cleared rapidly during labor and then delivery for the normal transition to air breathing to occur. Events that interfere with or delay this process cause respiratory distress, especially tachypnea.
Fluid in the Fetal Lung
Fetal lung fluid is actively secreted from alveolar type I and II cells at a rate of up to 20 mL/kg/d.24,25 This fluid is chloride rich and low in protein, the product of chloride secretion into the nascent airspaces after the creation of an electrochemical gradient by basolateral channels in epithelial cells.24,26 In the fetus, sodium follows chloride, and water follows the newly developed osmotic gradient to result in a net secretion of fluid by the fetal lung. Underlying mechanisms that allow the volume of fluid to be sensed and adjusted are still unknown, as is the identity of the primary chloride channel involved. What is clear is that this fluid is essential to normal lung growth and provides a distending volume similar to functional residual capacity (FRC). Processes that interfere with normal production and volume of fetal lung fluid, such as oligohydramnios and pulmonary artery ligation, result in pulmonary hypoplasia and respiratory distress.23,27
Newborn Transition
At the time of delivery after spontaneous labor, the transition from fluid-filled lungs to air-filled lungs has already begun. Animal studies have shown a decrease in the production of fetal lung fluid and net volume of fluid in the lungs occurs in the days prior to delivery.25,28,29 The initial stimulus for this process is still unknown, much as many factors in the onset of labor itself are still a mystery. Hormonal changes that accompany labor, including production of steroid hormones, catecholamines, thyroid hormone, and vasopressin, have all been shown to decrease the production of fluid by the fetal lung.25,28–30 With the addition of exposure to oxygen and mechanical stretch that follow delivery, the lung epithelium ultimately becomes a highly effective absorptive surface, with the majority of fetal lung fluid taken up into the pulmonary blood vessels and lymphatic system instead of out the mouth and trachea as the vaginal squeeze theory previously postulated.5
Studies performed on lung explants and fetal lambs revealed that sodium flux from the lung lumen to the plasma occurs, resulting in net lung fluid absorption.31,32 Sodium transport, and thus lung fluid resorption, is inhibited by administration of the diuretic amiloride,32 and administration to fetal rabbits resulted in heavier lungs, respiratory distress, and hypoxia at birth.33 The identification of ENaC, which is specifically sensitive to amiloride, helped to clarify further the essential role that secondary active sodium transport plays in the process.34 As shown in Figure 24-1, the basolateral Na+/K+ adenosine triphosphatase (ATPase) creates an electrochemical gradient that draws sodium into the cell through sodium channels that are newly present in the apical membrane. Anions and water follow, causing net fluid absorption. ENaC is responsible for 40% to 70% of the sodium resorption in the newborn period and has been extensively studied. Steroid hormones and catecholamines, shown to decrease the production of fetal lung fluid, have also been demonstrated to enhance ENaC activity and production,35–39 and serum cortisol levels were shown to correlate with α-ENaC levels in neonates.40
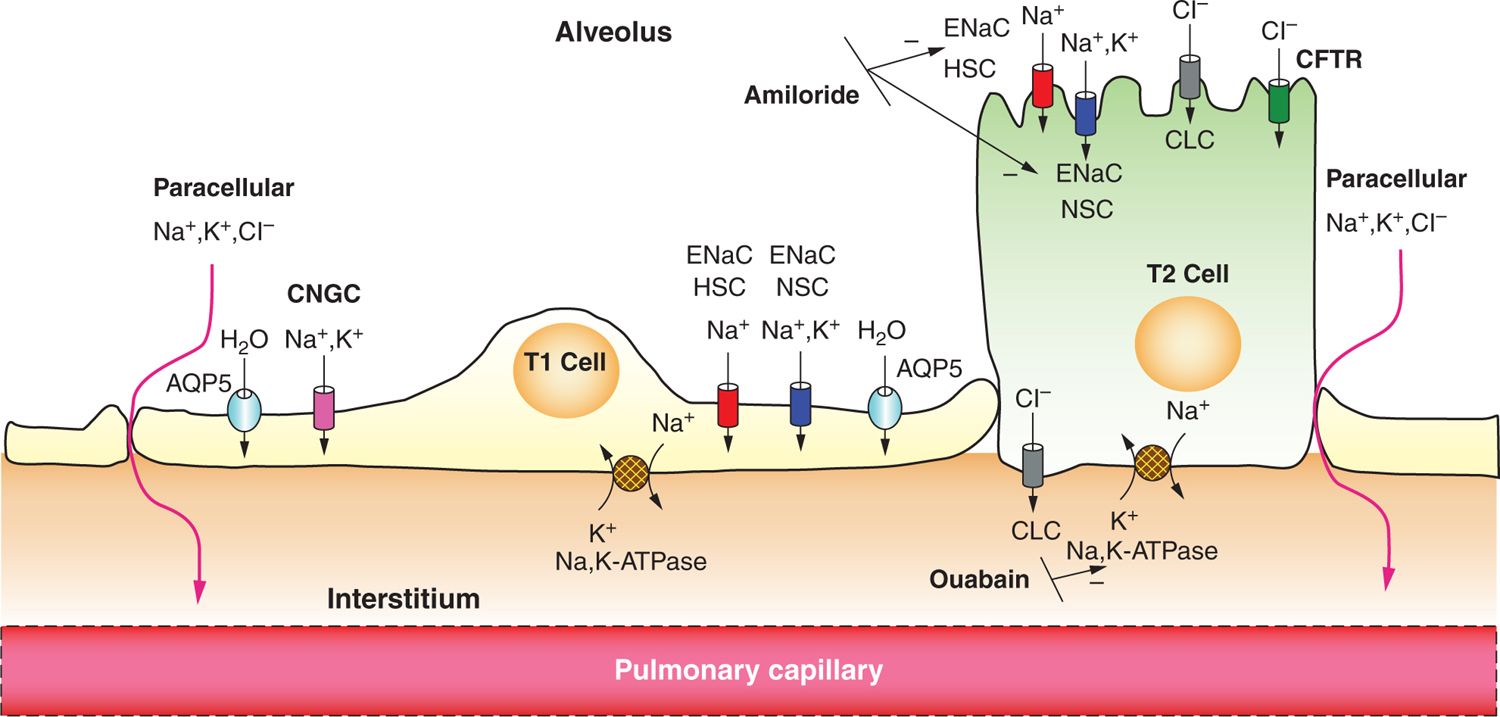
FIGURE 24-1 Epithelial sodium absorption in the fetal lung near birth. Near birth, Na+/K+ adenosine triphosphatase (ATPase) activity on the basolateral aspect of the cell membrane increases, which drives out 3 Na ions in exchange for 2 K ions, increasing the electrochemical gradient at the apical membrane. This process can be blocked by the cardiac glycoside ouabain. Na+ enters the cell through the apical surface of both alveolar type 1 (ATI) and type 2 (ATII) cells via amiloride-sensitive epithelial Na channels (ENaCs), both highly selective channels (HSCs) and nonselective channels (NSCs), and cyclic nucleotide gated channels (CNGC) (seen only in ATI cells). Electroneutrality is conserved with chloride movement through cystic fibrosis transmembrane conductance regulator (CFTR) or through chloride channels (CLCs) in ATI and ATII cells or paracellularly through tight junctions. Net ion movement is from the apical surface to the interstitium, creating an osmotic gradient, which in turn directs water transport in the same direction, either through aquaporins (Aquaporin-5, AQP5) or by diffusion.
There are 4 identified subunits of the ENaC channel: α, β, γ, and δ, with the role of δ-ENaC the least well known as it was only recently identified in respiratory epithelium.41 The α subunit has been the most studied, and its deficiency has been shown to cause respiratory failure that is neonatally lethal in mice; deficiency in the β and γ subunits does lead to an increase in lung water content but caused death of the mice from electrolyte abnormalities.36,42 The relative importance of the subunits is not clearly defined in humans, but all are necessary for normal function.39
Abnormalities in Transition
Nasal potential difference studies, which are performed on nasal epithelium as a surrogate for respiratory epithelium, indicated levels of sodium transport and thus ENaC activity. Support for the role of ENaC in the pathogenesis of TTN was demonstrated by Gowen et al in 1988; they showed that infants with TTN had a significantly higher nasal potential difference than healthy controls and did not show the typical decrease in response to amiloride, both indicative of low levels of ENaC.43 This was mirrored in infants delivered by cesarean section prior to the onset of labor, although they were often asymptomatic. Elevated respiratory rate and elevated nasal potential difference were correlated, and both decreased together over time and were equal to those of healthy controls at 72 hours of life.
It is clear that prematurely born infants are at increased risk for TTN. This is in part explained by evidence in animal and human studies that ENaC expression is developmentally regulated.31,35 Nasal potential difference studies in preterm infants with respiratory distress showed a smaller decrease in potential difference in response to amiloride than preterm infants without respiratory distress, indicating a lower level of ENaC activity.44 Further studies have confirmed quantatively lower levels of α-, β-, and γ-ENaC in preterm infants with respiratory distress when compared to healthy term infants.45,46 Trending levels of ENaC subunits over the first 30 hours of life in term infants delivered vaginally, term infants delivered via cesarean, and preterm infants have shown term infants have a decrease in ENaC levels as lung fluid is cleared, but this decrease is slower in those delivered by cesarean, as it is in premature infants.45 Thus, lower levels of epithelial sodium transport due to low levels of ENaC production, whether due to gestational immaturity, lack of spontaneous labor, or both, contribute to the development of TTN.
How retention of fetal lung fluid leads to such predictable tachypnea, in spite of usually normal or even low arterial CO2 levels, is a puzzling biological phenomenon. Early studies by Paintal et al47 suggested a role for pulmonary C fibers (J receptors) in initiating reflex activity leading to tachypnea and bronchoconstriction among other effects. Paintal postulated that C fibers respond to excess fluid in the interstitial space as interstitial stretch receptors triggering rapid shallow breathing, which may occur even in the presence of hypocapnia. A similar role has since been described for bronchial C fibers as well.48,49
CLINICAL PICTURE
The hallmark of TTN, as the name implies, is tachypnea appearing shortly after birth with respiratory rates greater than 60 breaths per minute (Table 24-1). Tachypnea may be accompanied by grunting, flaring, retractions, and cyanosis but is distinctly disproportionate to other signs of respiratory distress. Infants may have an increased oxygen requirement, but there is generally no hypercapnia. There is no clear consensus for the minimum duration of tachypnea needed to justify the label of TTN. Many experts recommend more than 6 hours of respiratory symptoms to differentiate the condition from the more transient delayed neonatal transition. Resolution of symptoms by 72 hours is seen in a majority of the infants. However, secondary complications such as pulmonary hypertension, acidosis, and air leak syndrome can alter the course and lead to severe hypoxic respiratory failure.50,51
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


