Neonatal Cholestasis
Cholestasis is biochemically characterized by conjugated or mixed hyperbilirubinemia, with elevated alkaline phosphatase and GGT, although some cases of PFIC and bile acid synthesis defects may have normal GGT. Any infant jaundiced beyond 10 days of age requires evaluation for cholestasis. The principal etiologic considerations in neonatal cholestasis include extrahepatic biliary atresia (EHBA), neonatal hepatitis, inherited/metabolic diseases, paucity of bile ducts, choledochal cyst, sepsis, drug-related injury, and total parenteral nutrition of some duration. The main metabolic and inherited diseases in the differential diagnosis are galactosemia, A1AT deficiency, PFIC, bile acid synthesis defects, and cystic fibrosis.
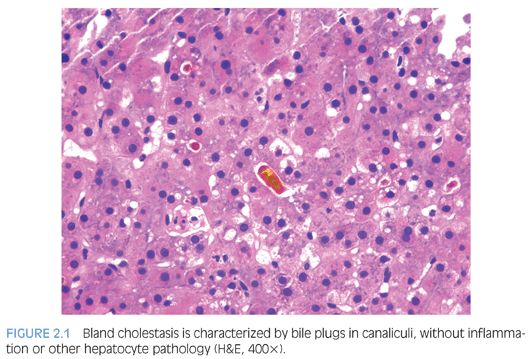
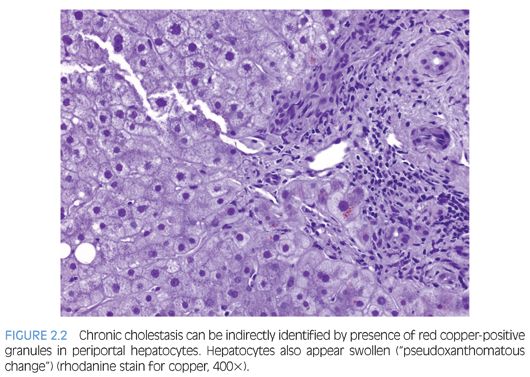
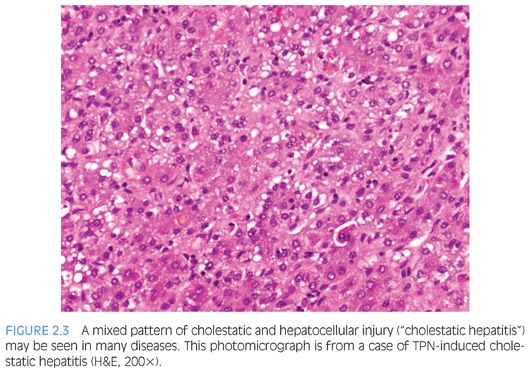
Analysis of general histologic patterns of cholestasis is helpful in overall biopsy evaluation. Histologically, cholestasis is characterized by the presence of bile pigment in hepatocytes, canaliculi, and/or Kupffer cells (Fig. 2.1). This is seen predominantly in centrilobular areas (acinar zone 3) in an acute setting, becoming more panlobular with increasing chronicity. With chronic cholestasis, there may be “pseudoxanthomatous” change of periportal hepatocytes due to cholate stasis. Early and mild cases of chronic cholestasis can be identified by a copper stain that highlights red granules in periportal (acinar zone 1) hepatocytes (Fig. 2.2). Although newborn infants may have stainable copper, a positive stain in older children is abnormal. In the presence of large duct obstruction and EHBA, there is accompanying portal tract reaction with edema, ductular proliferation, and bile plugging of ducts. Neutrophils may be seen in association with ductular reaction due to cytokine secretion, and their presence does not necessarily imply an infectious cholangitis. Lymphocytes and plasma cells may be present if obstruction has been present for weeks. In incomplete obstruction or if obstruction has been relieved by stenting, ductular reaction may be present without significant cholestasis. In the older child/adolescent, the presence of periductal onion skin fibrosis and fibroobliterative cholangitis should raise a possibility of primary sclerosing cholangitis (PSC). Unfortunately, children with PSC are often asymptomatic and do not manifest jaundice, leading to a delay in diagnosis.6 Many children with PSC may actually show morphologic features of autoimmune hepatitis. Cholestasis associated with active small bile duct damage is also seen in drug toxicity (e.g., Augmentin) and acute cellular rejection. Primary biliary cirrhosis does not typically occur in children. Chronic bile duct injury leads to ductopenia (loss of >50% bile ducts); again, one should look for ducts in the center of the portal tracts and compare caliber with hepatic artery branches to prevent mistaking ductules for ducts. In this setting, primary ductopenic diseases are in the differential, including syndromic and nonsyndromic bile duct paucity. Pure cholestasis without portal tract pathology may be seen in sepsis, drug toxicity, mutations of pericanalicular bile salt transport proteins, and in allograft ischemia-reperfusion injury. Cholestasis may accompany features of hepatocellular injury, including lobular disarray, hepatocyte ballooning and apoptosis, lobular and portal chronic inflammation, and presence of pigmented (ceroid) Kupffer cells (Fig. 2.3). This pattern of mixed hepatocellular and cholestatic injury (so-called cholestatic hepatitis) should raise a differential diagnosis of infections (e.g., hepatitis A, hepatitis E, cytomegalovirus [CMV], herpes simplex virus [HSV], rubella, parainfluenza, parvovirus, Listeria, syphilis, Toxoplasma), drug toxicity, autoimmune hepatitis, and metabolic diseases.
Extrahepatic Biliary Atresia
Identification of EHBA should be the first step in the evaluation of liver biopsy performed for neonatal cholestasis because this has major management implications. Although diagnosis of EHBA is possible by current imaging modalities, liver biopsy continues to have an essential role. Many biopsies are, however, performed today in an intraoperative setting, for frozen section diagnosis/confirmation. Most cases of EHBA are of the perinatal form and present at 4 to 8 weeks of age, after a jaundice-free period and without other associated anomalies. However, up to 35% of cases may present without a jaundice-free period (“fetal form”), and these may be associated with other congenital anomalies (e.g., polysplenia, asplenia, cardiovascular defects, abdominal situs inversus, intestinal malrotation, portal vein and hepatic artery anomalies).7
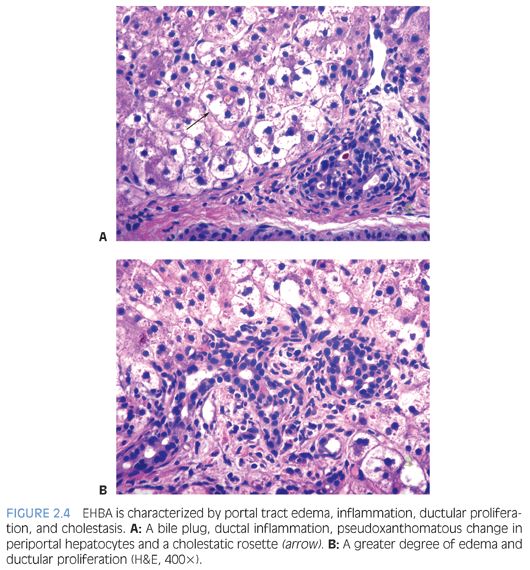
Histologically, EHBA is characterized by cholestasis, ductular proliferation, portal edema, and portal fibrosis (Fig. 2.4). As outlined earlier, cholestasis is most prominent in acinar zone 3. Bile plugs may be seen in ducts and ductules. Pseudoacini may form around canalicular bile plugs, so-called cholestatic rosettes. Ductular proliferation along with bile plugging is the salient diagnostic feature. Occasionally, however, ductular proliferation may occur only late in the course of disease leading to a delay in diagnosis.8 Ductular proliferation may be highlighted with a CK7 immunostain. A note of caution is that a CK7 immunostain in neonates also highlights persistent ductal plates, which may completely involute only in later infancy, and this can be mistaken for ductular proliferation. Persistent ductal plates are usually not associated with portal tract edema and inflammation. On the other hand, EHBA usually shows prominent periductal edema, and the ductal epithelium may show degenerative changes. With time, there is increasing portal fibrosis, progressing to biliary cirrhosis if left uncorrected. A mixed portal inflammation is often present. Severe cases may show absent bile ducts even in large interlobar portal tracts. Similar changes may be seen with large duct obstruction due to other causes (e.g., bile duct stenosis, choledochal cyst, cystic fibrosis). Unlike in adults with extrahepatic biliary obstruction, “bile lakes” (amorphous collections of bile surrounded by inflammatory cells and connective tissue) are rare. A1AT deficiency may mimic the histologic features of EHBA, and serum A1AT levels and phenotype should always be tested in these infants prior to surgery.9
Fragments of extrahepatic ducts may be sent for pathologic evaluation at or following surgery. These ducts display changes ranging from mild inflammation and epithelial dysmorphism to complete obliteration. The lining of relatively preserved larger ducts is often ulcerated, with intraluminal and extraluminal fibrosis distorting the lumen. At later stages, these ducts are often obliterated by fibrosis. The gallbladder may also be diminutive, exhibiting epithelial degeneration and fibrosis. In extreme cases, the liver may abut the duodenum, and the entire extrahepatic biliary tree may be missing.
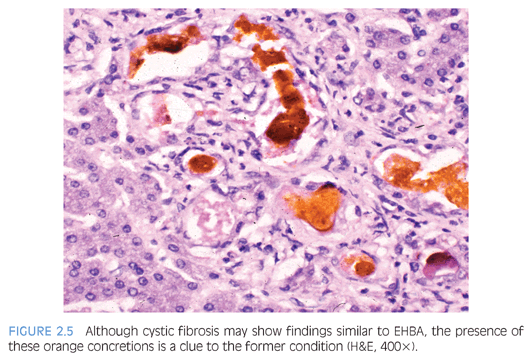
Although patients with cystic fibrosis tend to develop clinically significant liver disease beyond 10 years of age, transient neonatal or infantile cholestasis may occur in affected children associated with meconium ileus or mucus plugging.10 Histopathologic changes may mimic EHBA because both conditions have bile outflow obstruction. The pathognomonic lesion in cystic fibrosis is “focal biliary cirrhosis,” so-called due to its macroscopic appearance of focally depressed stellate scars. Proliferating and dilated ductules contain characteristic pink to orange concretions (Fig. 2.5), which may elicit inflammation as it spills out of ruptured ductules.11 Infants with cystic fibrosis may show portal fibrosis even in the absence of ductular reaction and inspissated secretion. With time, the foci of fibrotic biliary lesions coalesce, resulting in “multilobar cirrhosis,” resembling hepar lobatum.12
Neonatal Hepatitis
Neonatal hepatitis is a pattern of injury that may be seen in association with metabolic disease, infection, A1AT deficiency, PFIC, toxic injury, Alagille syndrome, hypopituitarism, autoimmune hemolytic anemia, and fetal thrombotic vasculopathy. Idiopathic neonatal hepatitis (INH) accounts for up to 40% of cases of neonatal cholestasis in some series and is a diagnosis of exclusion. Idiopathic cases are usually sporadic, although in some cases, there may be a family history of neonatal cholestasis, further underscoring the fact that they are not truly idiopathic but that the etiologic basis awaits elucidation. Sporadic cases have a more favorable prognosis (74% recovery) than those with familial forms (22% recovery).13
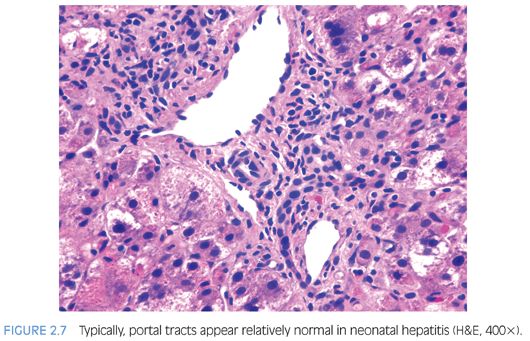
Hepatocyte giant cell transformation is usually prominent and panlobular (Fig. 2.6), but this by itself is not pathognomonic because it may be seen in many neonatal liver disorders, including metabolic diseases (Niemann-Pick, neonatal hemochromatosis), infections (CMV, rubella, herpes, parainfluenza), A1AT deficiency, PFIC, and Alagille syndrome. Although giant cell transformation may be also seen in EHBA, it is generally restricted to zone 1 hepatocytes at the interface of expanded portal tracts. Other hepatocyte abnormalities seen in INH include ballooning, apoptosis, and pseudoglandular or acinar formation. Lobular or portal inflammation is usually sparse14 and, if prominent, should suggest an infectious (e.g., viral) etiology. The main, and often also the most difficult, differential diagnosis is EHBA. The distinction is, however, important because EHBA requires early surgical correction (portoenterostomy/Kasai procedure). In most cases of neonatal hepatitis, unlike in EHBA, cholestasis is usually restricted to zone 3 hepatocytes and canaliculi and is rarely seen in the interlobular bile ducts. Furthermore, portal edema, expansion, and ductular proliferation are not the most prominent features of neonatal hepatitis (Fig. 2.7); in fact, bile ducts often appear hypoplastic.14
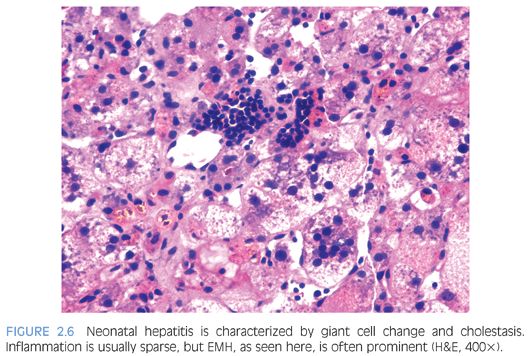
Extramedullary hematopoiesis (EMH) may be seen in both conditions and does not, by itself, aid differentiation. The presence of significant inflammation, fibrosis, ductular proliferation, and steatosis should alert the pathologist to other possible causes such as metabolic diseases and infections. In the absence of giant cells, etiologic considerations include Zellweger syndrome, Jeune syndrome, Alagille syndrome, CMV infection, neonatal lupus, and some forms of PFIC.8
Other Causes of Intrahepatic Cholestasis
Once the two main diagnostic categories of neonatal cholestasis have been considered, other less common etiologies deserve attention. Persistent intrahepatic cholestasis may be seen in inherited conditions or may be secondary to a variety of systemic causes such as infections (CMV, rubella, herpes, parainfluenza, gram-negative sepsis) and metabolic diseases (galactosemia, tyrosinemia, fructose intolerance, neonatal hemochromatosis, Zellweger syndrome, mitochondrial disorders).8 The main categories of inherited disorders in this situation include alterations in bile duct number (exemplified by paucity of bile ducts) and alterations in bile secretion (including PFIC and bile acid synthesis defects).
PAUCITY OF BILE DUCTS. Bile duct paucity may be syndromic (Alagille syndrome) or nonsyndromic. Causes of nonsyndromic paucity include sporadic cases of neonatal cholestasis with progressive liver disease, some cases of A1AT deficiency, Turner syndrome, Down syndrome, cystic fibrosis, hypopituitarism, medications, infections (e.g., CMV, hepatitis B virus [HBV], congenital syphilis), toxins, ischemic cholangiopathy, immune-mediated injury, and graft-versus-host disease.15,16
Alagille syndrome is an autosomal dominant disorder that occurs due to a mutation in JAG1 gene on chromosome 20p12 leading to abnormal notch signaling.17 Children with Alagille syndrome have characteristic facies (broad forehead, hypertelorism, flattened malar eminence, and pointed chin); ocular posterior embryotoxon; and cardiovascular, vertebral, and renal anomalies. Typically, cholestasis occurs in the first 3 months of life with unconjugated hyperbilirubinemia and an obstructive pattern on laboratory evaluation and hepatobiliary scintigraphy. Incomplete forms with late presentation have been described. Most patients tend to have mild disease and long-term survival.18
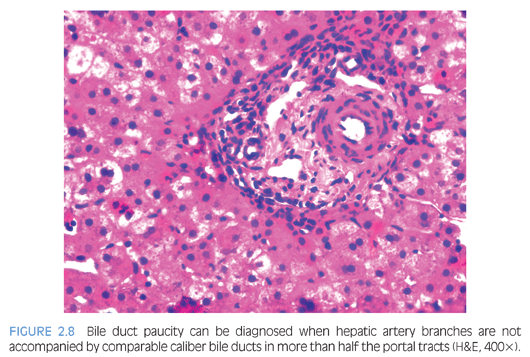
As the name suggests, the characteristic histologic feature of bile duct paucity is the absence or paucity of interlobular bile ducts, with bile duct to portal tract ratio of 0.4 or less (Fig. 2.8, eFig. 2.2). Bile duct paucity is progressive and may not be apparent until after 6 months of age. Intrahepatic (zone 3 dominant) cholestasis is usually present. Nonspecific features include hepatocellular ballooning, pseudoacinar transformation, focal giant cell formation, lobular disarray, and positive copper stains. Ductular proliferation and significant fibrosis are usually absent. Ultrastructurally, there are distinctive changes with bile pigment retention in the cytoplasm, especially in lysosomes and in vesicles in the outer convex region of the Golgi apparatus.19 Syndromic bile duct paucity is histologically indistinguishable from nonsyndromic paucity.
PROGRESSIVE FAMILIAL INTRAHEPATIC CHOLESTASIS (BYLER DISEASE AND BYLER SYNDROME). Originally described in Amish children, PFIC is a rare group of autosomally inherited disorders of bile acid transport. Bile acids help carry bilirubin into the canaliculus, and these defects therefore result in intrahepatic cholestasis. PFIC types 1 (ATP8B1 gene mutation at 18q21) and 2 (ABCB11 gene mutation at 2q24) are characterized by cholestasis and low serum GGT activity, whereas PFIC type 3 (ABCB4 gene mutation at 7q2) is associated with elevated serum GGT. The currently preferred nomenclature for the three PFIC disorders is familial intrahepatic cholestasis (FIC1) deficiency, bile salt export pump (BSEP) deficiency, and MDR3 deficiency respectively.20 In PFICs, serum bile acid levels are elevated. Patients present with conjugated hyperbilirubinemia and progress to cirrhosis.
Histologic features are variable and depend on the stage of progression to cirrhosis. Amish children with PFIC type 1 typically exhibit bland cholestasis and bile duct paucity, without significant lobular pathology or fibrosis in the first 6 months of life. With progression, there is giant cell transformation, pseudoacinar change, and progressive spidery fibrosis, the latter beginning in zone 3 and extending to zone 1, eventually leading to cirrhosis. Non-Amish children have neonatal hepatitis and a more benign course but with recurrent cholestasis. PFIC type 2 is also characterized by persistent neonatal cholestasis with features of neonatal hepatitis but progress to biliary cirrhosis. PFIC type 3 displays periportal inflammation and extensive bile duct proliferation and may thus mimic EHBA. Immunohistochemical stains have been developed toward BSEP and MDR3 proteins to identify lack of expression in PFIC2 and PFIC3, respectively. Ultrastructurally, Amish children with PFIC1 have coarse granular bile, whereas non-Amish children and other types may have amorphous to finely filamentous bile. Histologic overlap, with normal GGT levels, may also be seen in hypopituitarism and bile salt synthesis defects.
BILE ACID SYNTHESIS DEFECTS. Similar to PFIC, patients with bile acid synthesis defects also present with neonatal cholestasis and normal serum GGT levels. Urinary bile acid levels are elevated. These autosomal recessive conditions are usually diagnosed by mass spectrometry of bile acids in urine or bile, and patients are rarely biopsied. Histologic features resemble neonatal hepatitis, with prominent cholestasis and bridging fibrosis. Ultrastructurally, bile canaliculi may appear abnormal and dense granular bile residue may be seen.
Other conditions associated with intrahepatic cholestasis include benign recurrent intrahepatic cholestasis and Aagenaes syndrome (hereditary cholestasis with lymphedema). The former is characterized by recurrent episodes of cholestasis without permanent liver damage.21 The latter presents with cholestasis at birth, high serum GGT levels, and lymphedema; cholestasis may be recurrent and tends to improve with age. Crigler-Najjar disease presents with unconjugated hyperbilirubinemia in the first 3 days of life, with other routine liver tests being within normal range. Liver biopsy may show intrahepatic cholestasis or may be normal.22 Gilbert syndrome usually has mild intermittent unconjugated hyperbilirubinemia beginning in adolescence; again, liver enzyme levels are normal. Dubin-Johnson and Rotor syndromes usually present in adult life with conjugated hyperbilirubinemia.
Alpha-1 Antitrypsin Deficiency
A1AT deficiency is an autosomal recessive disease caused by mutations in the protease inhibitor gene (Pi) on chromosome 14q, leading to unopposed neutrophil elastase activity, and characterized by liver and lung pathology. Mutant A1AT is unable to fold properly, resulting in failure of translocation from the endoplasmic reticulum to the Golgi apparatus. This mutant A1AT then accumulates in the hepatocyte, leads to hepatocyte injury, and presents variably with neonatal cholestasis in infancy to recurrent hepatitis in young adults, with progression to chronic hepatitis and cirrhosis. It accounts for over 10% of cases of neonatal cholestasis, making it the most common genetic cause of neonatal liver disease.23
Characteristic histology in the neonate includes cholestasis, pseudoacinar and giant cell transformation, and EMH. The degree of portal fibrosis and ductular proliferation are variable, with some cases resembling EHBA. Paucity of bile ducts may be seen. Extensive hepatocellular necrosis may occur with resultant clinical fulminant hepatic failure. The well-known diastase-resistant periodic acid–Schiff (PAS)–positive eosinophilic hyaline globules are seen mainly in zone 1 hepatocytes (eFig.2.3A) and occasionally in bile duct epithelium. One caveat is that these globules may not be seen in biopsy specimens obtained in the first few months of life. However, even in this setting, the stored material is often demonstrable by immunohistochemistry (eFig. 2.3B). On electron microscopy, these globules are flocculent and moderately electron-dense, present within dilated cisternae of rough endoplasmic reticulum. The definitive diagnosis of A1AT deficiency is, however, made on serum quantitation and phenotype studies.
Total Parenteral Nutrition–Related Injury
The association of total parenteral nutrition (TPN) with hepatic dysfunction is well known.24 Patients typically present with cholestasis. Prematurity, low birth weight, and low gestational age are the greatest risk factors. The injury is also more common and more severe in infants with gastrointestinal disease, intestinal resection, and with longer duration of TPN. Affected infants may only manifest an insidious onset of jaundice. Monitoring serum bile acid levels is helpful in early identification because elevated serum bile acid is the earliest biochemical abnormality, beginning as early as 5 days after initiating TPN. Hyperbilirubinemia, on the other hand, may be seen only after 3 or more weeks of treatment. Most patients recover after recommencement of enteral feeding. The diagnosis remains one of exclusion.
Histologically, there is evidence of both cholestatic and hepatocellular injury (see Fig. 2.3). The initial and constant feature is intrahepatic cholestasis. As in other causes of cholestasis, this is most pronounced in acinar zone 3. Features of hepatocellular injury include lobular disarray, ballooning, apoptosis, giant cell transformation, Kupffer cell hyperplasia, and pigmented Kupffer cells (as evidence of hepatocyte injury). These scavenger cells may be highlighted by a periodic acid–Schiff diastase (PASD) stain. EMH may be prominent, and inflammation is of variable severity. Lymphocytes predominate in the inflammatory infiltrate, although eosinophils and neutrophils may also be present. Portal edema, pericholangitis, and fibrosis (both portal and perisinusoidal) may be seen. Histologic recovery is usually complete, although features of injury may persist, as in any chronic liver disease.
METABOLIC DISEASES
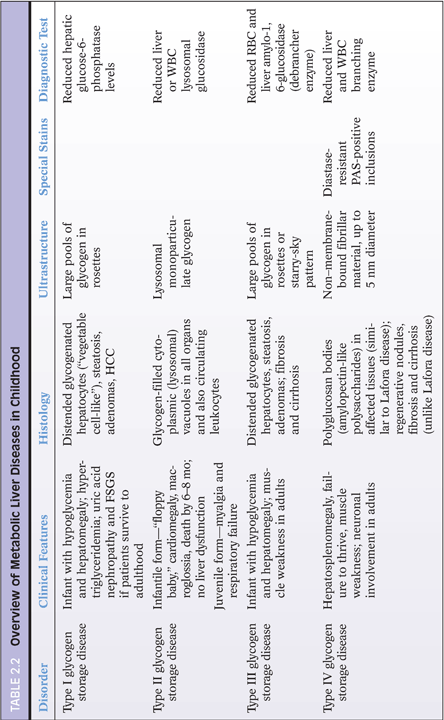
Metabolic diseases may mimic all patterns of liver injury in the pediatric age group (see Table 2.1). In neonatal life, metabolic disorders present with neonatal cholestasis and are therefore an important consideration. In later childhood, metabolic diseases are usually clinically suspected. Most metabolic diseases are diagnosed by biochemical and enzymatic investigations, and histology is often not diagnostic. However, histology certainly provides direction and may be the first clue to the diagnosis. A detailed description of hepatic morphology in various metabolic diseases is beyond the scope of this chapter; several excellent reviews are available for the interested reader.25–28 An overview of liver histopathology in various metabolic diseases is given in Table 2.2. Select examples are illustrated in Figures 2.9 to 2.15.