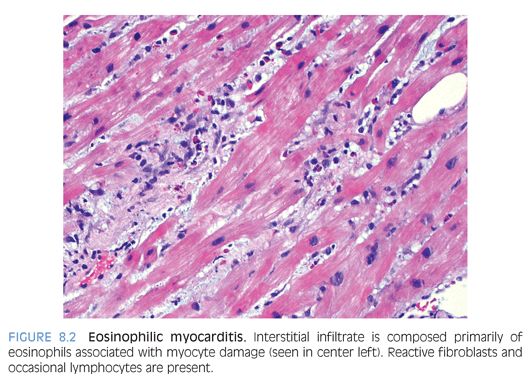
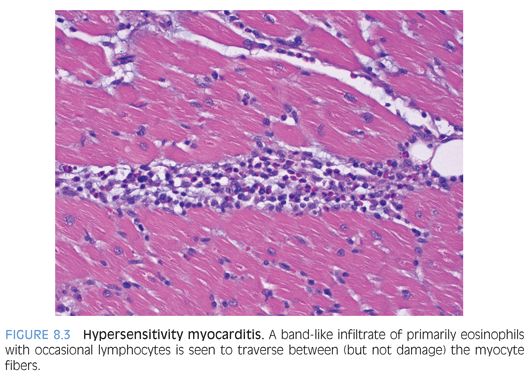
Eosinophilic inflammation: Eosinophilic (Löffler) myocarditis is a rare form of myocardial inflammation associated with eosinophilia, which is most commonly idiopathic but can be seen in parasitic infection, hypereosinophilic syndromes, asthmatic bronchitis, Mycoplasma pneumonia, and malignancy.9 Eosinophils comprise the majority of inflammatory cells with a few lymphocytes, plasma cells and macrophages (Fig. 8.2), and myocyte necrosis. Extensive eosinophil degranulation may result in endocardial damage, seen as attached fibrin with entrapped eosinophils. Hypersensitivity myocarditis is usually drug-induced and is characterized by a prominent interstitial eosinophilic infiltrate (Fig. 8.3) and only a few lymphocytes, plasma cells, macrophages, and rare giant cells. The hallmark is asynchrony between the severity of the infiltrate and little, if any, myocyte necrosis, with ultimate resolution of the infiltrate and return to normal cardiac function upon withdrawal of the offending agent.10
Giant cell myocarditis is a rare, aggressive disease of unknown etiology that is characterized by a mixed chronic inflammation with prominent component of giant cells11 but no well-formed granulomas or myocyte necrosis. Although predominantly an adult disease, it does occur in the pediatric age range, predominantly the second decade. Because approximately 20% of patients have an underlying autoimmune disorder, especially inflammatory bowel disease, an immune-mediated pathogenesis is presumed.6
Sarcoidosis-associated myocarditis is rare in children.12 Well-formed nonnecrotizing granulomas replace patches of myocardium. Giant cells are often present and zone of inflammation may be extensive and confluent. In later stages, especially after steroid therapy, fibrosis may be prominent.
CARDIOMYOPATHY
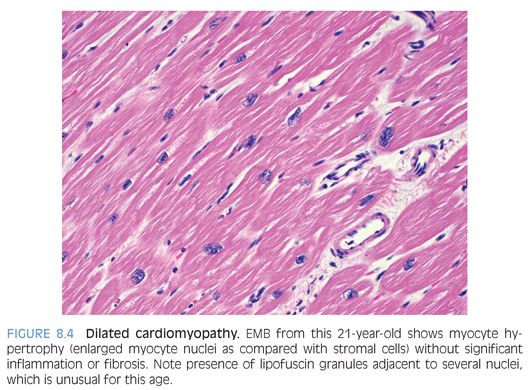
Dilated cardiomyopathy is characterized by contractile dysfunction of the myocardium that results in decreased ejection fraction; dilatation of cardiac chambers; and diffuse ventricular hypokinesis with no systemic etiology such as valvular/congenital heart disease, pulmonary disease, or hypertension.13 About one-third may be secondary to genetic defects. Microscopic findings are nonspecific and include interstitial or focal fibrosis, myocyte hypertrophy (best identified by nuclear enlargement) with concomitant myofibrillar loss, or myocyte atrophy (Fig. 8.4). Lipofuscin pigment is prominent and there may be a mild chronic lymphocytic inflammation. EMB is performed mainly to rule out myocarditis.
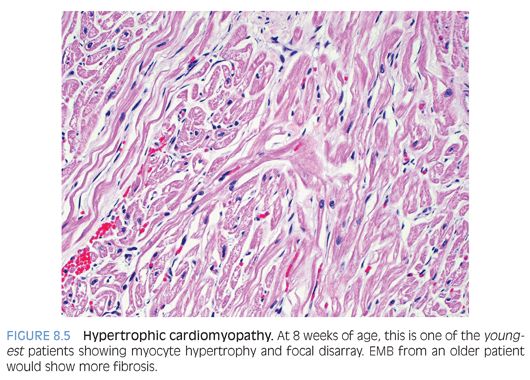
Hypertrophic cardiomyopathy (HCM) is a primary disorder of cardiac muscle that leads to asymmetrical myocardial hypertrophy classically involving the interventricular septum,14 which should be at least 1.5 times the thickness of the posterior left ventricular wall. Mutations in the β-cardiac myosin have been found in about half of the cases. There is myocardial fiber disarray (Fig. 8.5) characterized by disorganized, whorled, or tangled orientation of myocardial fibers that are multipolar (i.e., with prominent branching). There is myocyte hypertrophy, interstitial fibrosis, and thickened intramyocardial arterioles. It should be noted that small areas of disarray may occur in normal hearts, especially at the junction of the interventricular septum and right ventricle.
Arrhythmogenic right ventricular dysplasia/cardiomyopathy is characterized by arrhythmia and right heart dilatation with focal replacement of myocardium by fat and fibrous tissue. Mutations in desmosomal genes have recently been shown to be causative.15 The left ventricle is also involved in about one-fifth of the patients. There is loss of myocytes with intermingling of fat, fibrous tissue, and myocytes. Normal fat infiltration of the right ventricle is seen in obese patients and may be difficult to distinguish on biopsy.
Endocardial fibroelastosis (EFE) is a proliferation of fibroelastic tissue beneath the endocardium, which may extend into the underlying myocardium of any chamber. Rarely, EFE is a primary cardiomyopathy when it mainly involves the left ventricle. More often, it is seen in association with congenital cardiac malformations and other cardiomyopathies. In one study, EFE accounted for 25% of pediatric cases transplanted for dilated cardiomyopathy.16 Because mumps or adenoviral infections are identified in the majority of patients, an in utero viral infection is thought to be etiologically relevant.
Cardiac hemochromatosis may be due to autosomal recessive hemochromatosis, hemosiderosis due to chronic transfusion for anemia (thalassemia or sickle cell anemia), or rarely due to chronic ingestion of iron salts. Juvenile hereditary hemochromatosis occurs due to mutations in hemojuvelin (HJV, 1q21)17 or hepcidin (HAMP, 19q13.1) genes.18 Intramyocellular iron deposits are present and can be confirmed by iron staining, EM, or biochemical analysis. Myocellular degeneration is more obvious in the subepicardium rather than the subendocardium, and there is relative sparing of atria and conduction system. Increased iron may also accumulate in patients with Wilson disease (copper toxicity).
Dystrophy-associated cardiomyopathy (Duchenne muscular dystrophy, DMD) is a sex-linked recessive disorder that results in absence of dystrophin, a high-molecular-weight protein normally present within the sarcolemma of skeletal muscle.19 There is severe subepicardial fibrosis of the left ventricle but the subendocardium also shows areas of fibrosis with interspersed myocytes. Characteristically, there is no inflammation in the zones of fibrosis. Ultrastructurally, there is evidence of myofibrillar loss with disorganized Z-band material and preservation of transverse tubules.
Drug cardiotoxicity can occur with many cytotoxic drugs20 of which Adriamycin is the most common.21 Myocyte necrosis and extensive vacuolization may be seen on EMB. The diagnosis is often made clinically and EMB (when performed) is to rule out other etiologies.
METABOLIC/STORAGE DISORDERS
These are mentioned only briefly because the diagnosis is usually made on clinical, molecular genetic, and enzymatic tests, and the heart is only rarely biopsied.
Glycogen storage diseases are caused by a deficiency in one or more of the enzymes involved in the synthesis or degradation of glycogen, leading to the accumulation of glycogen in various tissues. Cardiac involvement is almost ubiquitous in type II (Pompe disease), whereas there is variable cardiac involvement in type III (Cori disease) and type IV (Anderson disease). Glycogen excess within the myocytes leads to mechanical cardiac failure.
Mucopolysaccharidoses are a family of hereditary diseases characterized by the accumulation of mucopolysaccharides due to deficiency in a lysosomal enzyme normally responsible for their degradation. These conditions lead to changes in cardiac valves, skin, cartilage, and bone, presumably due to the effect of acid mucopolysaccharides on collagen. Mucopolysaccharidosis I is characterized by reduced activity of α-L-iduronidase; excessive urinary secretion of dermatan sulphate and heparan sulphate; and the presence of large, oval, or rounded connective tissue cells (Hurler cells) within the valves, endocardium, myocardium, coronary arteries, and aorta. These cells are filled with numerous clear vacuoles containing acid mucopolysaccharide material. Molecular genetic investigation can reveal the genetic defect, which is located on the short arm of chromosome 4 (4p16.3).22 Mucopolysaccharidosis II or Hunter syndrome is an autosomal recessive disorder characterized by an enzymatic defect in iduronate-2-sulfatase. Cardiovascular disease is part of the spectrum for the majority of patients with either the mild or the severe form of disease. Microscopically, valves reveal the presence of large, clear cells similar to those seen in patients with Hurler syndrome (earlier discussion) with increased fibrous tissue. Clear cells are also present in the myocardium and endocardium but not in coronary arteries. Molecular genetic investigation can reveal the genetic defect on the X chromosome (Xq27.3).23 Mucopolysaccharidosis III or Sanfilippo syndrome has only a few stigmata other than mental retardation. The heart is morphologically involved. Microscopically, intracellular deposits are localized in small granular cells, but large Hurler-like cells are not always described. Mucopolysaccharidosis IV or Morquio syndrome has two biochemically distinct autosomal recessive forms. Cardiac involvement is well recognized. Mucopolysaccharidosis VI or Maroteaux-Lamy syndrome is characterized by an infiltration of foam cells with ultrastructural evidence of parallel electron-dense lamellae.
Carnitine deficiency–associated cardiomyopathy is characterized by low plasma carnitine levels in a child with biventricular heart failure. Myocytes are enlarged and contain lipid vacuoles that can be confirmed with special stains for lipids. On EM, there is disruption of the myofibrils and aggregation of mitochondria which have bizarre shapes and twisted cristae.
Sphingolipidosis: Fabry (Anderson-Fabry) disease is an X-linked inborn error of glycosphingolipid metabolism (3p21-23). Cardiac disease manifests most commonly as left ventricular hypertrophy.24 Microscopically, cardiac muscle cells reveal glycosphingolipid deposits that occupy the central and perinuclear zones, displacing contractile elements toward the periphery.
HEART TRANSPLANTATION
Pediatric heart transplantation is increasingly being performed for congenital heart disease without pulmonary hypertension, cardiomyopathy, valvular heart disease, or intractable arrhythmias. Posttransplant EMB plays an integral role in recipient management for which excellent surveillance biopsy protocols have been established by the International Society of Heart and Lung Transplantation (ISHLT).25 To be considered adequate, at least three fragments of myocardial tissue with the myocardium occupying more than 50% of the tissue in the biopsy fragment should be present for evaluation.25 Approximately 70% of biopsies are negative for rejection.
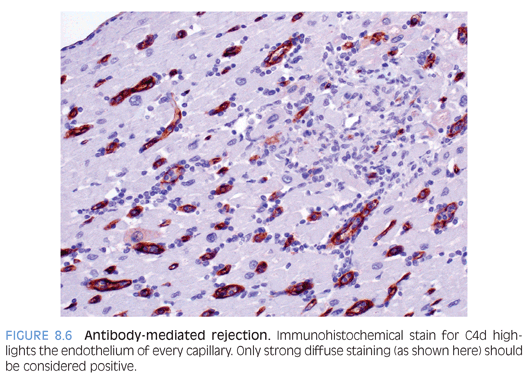
Antibody-mediated rejection (AMR) is a form of immune-mediated rejection that occurs due to formation/presence of circulating antibodies in the recipient to the donor graft. The incidence is variable, depending on a variety of factors, but is estimated to be between 10% and 20%.26 It may present within a few hours of transplantation or months to years later (most common in the first month). Clinically, there is evidence of hemodynamic compromise (hypotension, shock, decreased cardiac output). AMR may persist for several months with an associated poor outcome. Microscopically, AMR is characterized by endothelial swelling, interstitial edema, and only minimal inflammatory infiltrate. Intravascular thrombi may be noted. Morphologic findings are nonspecific and strong diffuse immunoreactivity (by IHC or IF) for complement factors C4d (Fig. 8.6) and/or C3d aids in the diagnosis.25 The main differential diagnosis is acute cellular rejection, which does not stain for C4d or C3d, as discussed below.