BONE MARROW
Considerations on the Normal Marrow
What is “normal” for a pediatric patient? The bone marrow is very dynamic in childhood, as reflected by the changes in hematologic values in the complete blood count. The expected proportions of the lymphoid, myeloid, and erythroid components vary, and those components may appear more “left shifted” with relatively more immature forms. This is especially true for lymphocytes and megakaryocytes and important to recognize, especially when evaluating for possible myelodysplasia, leukemia, or tumor staging.
At birth, all marrow cavities contain red hematopoietic marrow, which is gradually replaced by fat as the child grows. Marrow cellularity gradually declines with age, and although the maxim of using 100-age to estimate normal cellularity works fairly well in adults, this is not a good yardstick to use in childhood because the ranges are lower than that formula would suggest and fairly broad. At birth, marrow cellularity may approach 100% and is 90% to 100% in the neonatal period. This declines slightly to 80% to 90% in infancy, with ranges of 60% to 80% expected in early childhood and 50% to 70% after the age of 6 years.
As cellularity changes over time, so does the composition of the marrow. Although rarely biopsied, at birth, there is a relative predominance of myeloid elements with some degree of left shift and up to 5% blasts. This rapidly changes as the number of lymphocytes increases in the neonatal period, primarily consisting of hematogones or maturing B cells. In fact, the number of lymphocytes can be quite striking and the dominant cellular component, making the distinction between erythroid precursors and lymphocytes challenging on biopsy or clot sections. The myeloid to erythroid ratio also changes over time, with a high myeloid to erythroid ratio in the realm of 5:1 or higher during infancy and early childhood, especially during the physiologic erythroid nadir in the first few months of life and gradually approaching more adult levels (3 to 4:1) by the age of 6 years. Because children are rapidly growing and developing, features of bony remodeling are often visible on bone marrow biopsy, and osteoblasts and osteoclasts may be readily identified on bone marrow aspirate smears, especially in very young children. Lastly, although absent iron stores in an adult patient is diagnostic of iron deficiency, children younger than the age of 4 or 5 years do not have stainable iron stores.
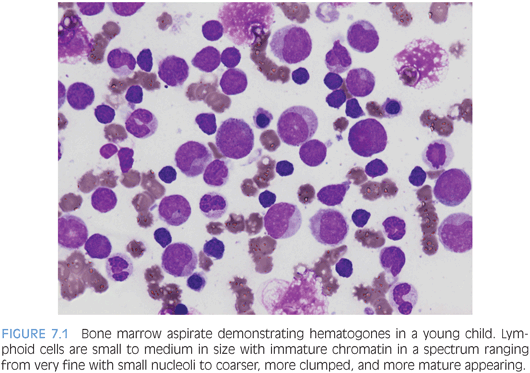
As the lymphoid component differs considerably between children and adults, this will be discussed in further detail. Unlike adults, who commonly demonstrate lymphoid aggregates, the lymphoid population in children is more evenly dispersed through the marrow in an interstitial pattern and composed largely of B cells in various stages of maturation (hematogones). T cells tend to appear in very small aggregates, often only visible by immunohistochemical studies. This is important to keep in mind because the number of B cells may appear alarming to someone used to looking at adult bone marrows. On aspirate smears, hematogones are small to intermediate in size with higher nuclear to cytoplasmic ratios and more immature appearing dispersed chromatin compared to mature lymphocytes (Fig. 7.1). Unlike the lymphoblasts of ALL, hematogones should have a spectrum of forms in various stages of maturation, and although the nuclear to cytoplasmic ratio is very high, it is even higher in lymphoblasts that have a more “clonal” appearance. Similar to the morphologic spectrum seen in hematogones, likewise, they demonstrate a maturation spectrum in their immunophenotype with variable expression of CD10, CD20, and surface immunoglobulin. Lastly, plasma cells are less frequently seen in the pediatric marrow compared to adults.
Reactive and Infectious
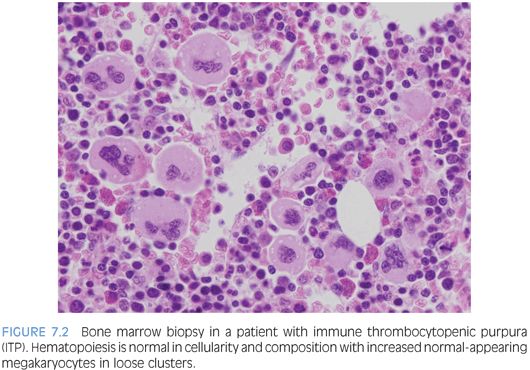
IMMUNE THROMBOCYTOPENIC PURPURA (ITP). ITP is commonly seen in children and usually presents as a self-limiting condition after a viral prodrome with easy bruising and petechiae. Bone marrow biopsy is infrequently performed for routine cases. In general, a bone marrow is performed if the patient has chronic ITP, additional cytopenias, or other reasons to suspect a possible marrow failure disorder or leukemia. The bone marrow will show increased numbers of megakaryocytes, which appear in loose clusters and may show some left shift with increased immature forms, recognized by their decreased lobation, higher nuclear to cytoplasmic ratio, and cytoplasm ranging from being deeply basophilic and relatively agranular to demonstrating a partial or complete complement of platelet-specific granules2 (Fig. 7.2). The marrow cellularity, myeloid, erythroid, and lymphoid components, will appear normal.
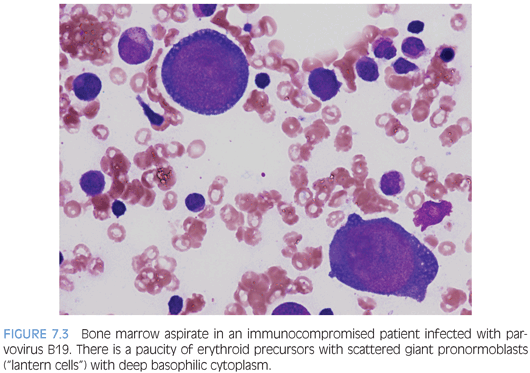
INFECTIONS. A variety of viral, fungal, or parasitic pathogens may infect the bone marrow. Many will come to clinical attention due to fevers and systemic symptoms or consequences of cytopenia affecting one or more cell lines. Viral infections, in particular, may cause some degree of marrow suppression, but whether or not they become clinically apparent depends in part on the immune status of the host and whether or not an underlying primary hematologic disorder is present. A bone marrow exam may be performed in these cases to collect specimens for microbiologic cultures and molecular studies for organisms as well as for morphologic exam. Some agents, such as mycobacteria, fungi, or leishmaniasis may be visible on aspirate smears or bone marrow biopsy and may be highlighted with the use of special stains. Most viral pathogens do not induce specific morphologic features in the bone marrow, although lymphocytosis with reactive “atypical” lymphocytes may be seen in the peripheral blood. Parvovirus B19 infection is distinct and, with the characteristic morphologic features, most prominent and easily appreciated in an immunocompromised host. Giant erythroid pronormoblasts are seen on bone marrow biopsy with vesicular chromatin and a large prominent central eosinophilic nucleolus (eFig. 7.1). On aspirate smears, these pronormoblasts have deeply basophilic cytoplasm sometimes with a few cytoplasmic vacuoles (the so-called lantern cells) (Fig. 7.3). Some may show intranuclear inclusions that are vaguely demarcated. Background myeloid, megakaryocytic, and lymphoid components are normal in number and appearance.
Bone Marrow Failure
Bone marrow failure is an uncommon cause of cytopenias and may be classified according to the number of cell lines affected and whether the condition is inherited or acquired. Because most inherited causes of single or multilineage marrow failure present in childhood, all patients presenting with marrow failure not attributed to an obvious acquired cause should be worked up for an inherited condition. Morphologic features alone do not distinguish between inherited constitutional or acquired conditions, and so correlation with history, physical, and other laboratory studies, often including genetic testing, is required. It is particularly important to identify these patients because many have other associated conditions or an increased risk for malignancy and require clinical monitoring and consideration for different conditioning regimens should they require bone marrow transplant.3
BONE MARROW FAILURE ASSOCIATED WITH SINGLE CYTOPENIAS
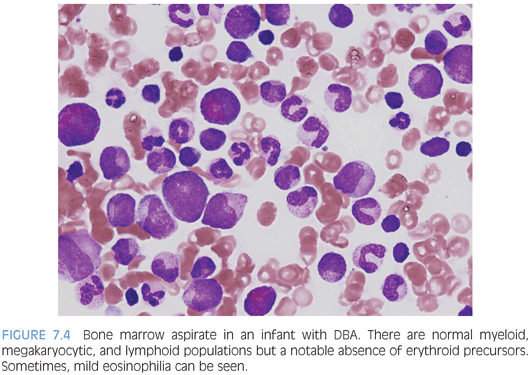
PURE RED CELL APLASIA. Diamond-Blackfan anemia (DBA) is the primary cause of inherited pure red cell aplasia and is autosomal dominantly inherited, although a large percentage will prove to be new mutations. It presents in the first year of life with severe macrocytic anemia and low reticulocyte counts. Other characteristic features, when present, include congenital anomalies (especially skeletal anomalies involving the thumb), elevated red cell adenosine deaminase (ADA), and elevated fetal hemoglobin (Hgb F), although the latter two features may be difficult to interpret in posttransfusion samples or due to the patient’s young age (Hgb F). Mutations in various genes encoding ribosomal subunits have been described in approximately half of patients with DBA, with the remainder of patients having as of yet undiscovered mutations. The bone marrow aspirate and biopsy findings show normal marrow cellularity with profound erythroid hypoplasia but otherwise essentially normal myelopoiesis and megakaryopoiesis with a normal lymphoid component. Mild eosinophilia may also be seen (Fig. 7.4).
Acquired pure red cell aplasia may be seen in the setting of parvovirus B19 infection (discussed earlier), transient erythroblastopenia of childhood (TEC), secondary to a drug (particularly noted with azathioprine), or thymoma. The main differential diagnosis with DBA is TEC. TEC is a self-limited disorder of unknown etiology that usually presents in the first few years of life with profound anemia and low reticulocyte count and spontaneous recovery within a month or two of presentation. The erythrocyte ADA levels are normal in TEC, and congenital abnormalities are absent. Otherwise, the morphologic and laboratory features of TEC overlap with DBA, so careful clinical correlation is necessary.
ISOLATED NEUTROPENIA. A variety of inherited disorders may present with neutropenia. These include severe congenital neutropenia, Shwachman-Diamond syndrome (SDS), Barth syndrome, and cyclic neutropenia. Neutropenia may also be a presenting or characteristic feature of certain multilineage marrow failure disorders, immunodeficiencies, storage diseases, or syndromes associated with oculocutaneous albinism (e.g., Chediak-Higashi syndrome). Acquired neutropenia in childhood is most commonly due to immune causes and antineutrophil antibodies but may also be seen secondary to medications, infections, bone marrow infiltration, or nutritional deficiencies.
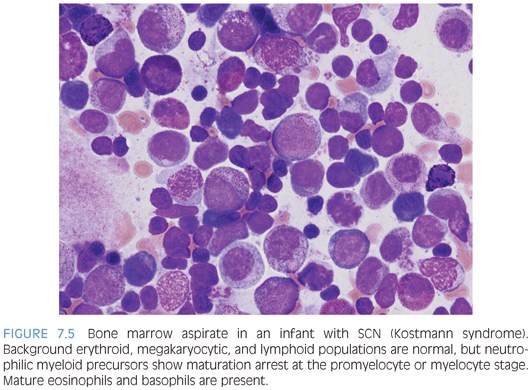
Severe congenital neutropenia (SCN) encompasses several disorders, which all share severe persistent isolated neutropenia and arrested myelopoiesis at the promyelocyte or myelocyte stage. Peripheral counts also usually show eosinophilia and monocytosis but normal red blood cell and platelet counts. Inheritance may be autosomal recessive or autosomal dominant, and several different genes have been implicated, including ELA2, HAX1, GFI1, and G-CSFR. Kostmann syndrome is part of the SCN spectrum and is used to refer to the autosomal recessive type.4 The bone marrow typically shows normal or mildly decreased cellularity for age. Myeloid precursors are present with a maturation arrest at the promyelocyte or myelocyte stage, and these may have abnormal dyspoietic features (Fig. 7.5). More mature neutrophils or neutrophilic precursors will be absent in untreated SCN, and eosinophils and eosinophilic precursor are relatively increased. Other cell lines appear normal in number and morphology.
Cyclic neutropenia is an autosomal dominant disorder associated with mutations in the ELA2 gene and 21-day cycles of neutropenia that is severe at the trough and then normal or mildly decreased at the peak. Fever and infections will follow the neutrophil counts at the troughs. Mild anemia and thrombocytopenia may accompany the severe neutropenia. The bone marrow features will vary with the neutrophil counts, with decreased myelopoiesis and maturation arrest to more normal myeloid development. Following weekly neutrophil counts over time for 1 or 2 months should establish the cyclical pattern and aid in the diagnosis.
ISOLATED THROMBOCYTOPENIA. There are a variety of inherited causes of thrombocytopenia and platelet abnormalities, which are further classified on the basis of platelet size because some disorders are associated with large-, normal-, or small-sized platelets. The thrombocytopenia or platelet abnormality presents as either the dominant feature of the disorder or one of many components of a syndrome associated with other clinical and laboratory features. The most common causes of acquired thrombocytopenia are medications and ITP (discussed earlier).
Macrothrombocytopenias are characterized by low platelet counts with consistently large platelets and represent a heterogenous group of disorders. Some are associated with bleeding complications either due to the thrombocytopenia or defects in platelet function. The more notable of these disorders are the MYH 9–related disorders (May-Hegglin being the most well known) and Bernard-Soulier syndrome. There is also the more recently described X-linked thrombocytopenia with dyserythropoiesis associated with GATA1 mutations. For the most part, bone marrow exams are not performed for these entities, other than latter entity related to GATA1 mutations.
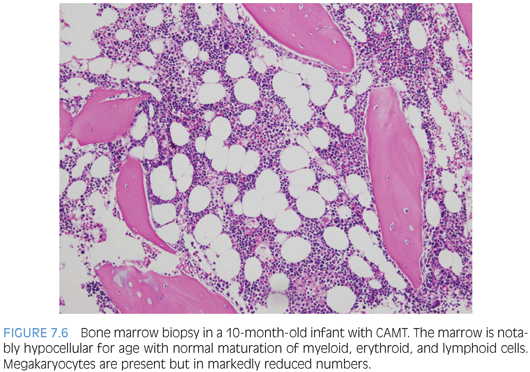
Inherited thrombocytopenias associated with normal cell size include congenital amegakaryocytic thrombocytopenia, thrombocytopenia with absent radii, and familial platelet disorder with associated myeloid malignancy associated with RUNX1 abnormalities. Congenital amegakaryocytic thrombocytopenia (CAMT) is an autosomal recessive disorder due to mutations in the MPL gene and is characterized by variable thrombocytopenia that is persistent and present at birth, absent megakaryocytes in the bone marrow, and progression to multilineage bone marrow failure in a large subset of patients. Bone marrow biopsy shows bone marrow that appears otherwise normal in cellularity and composition with a notable decrease or absence of megakaryocytes (Fig. 7.6). The severity and timetable to development of multilineage bone marrow failure varies with the type of MPL mutation and may occur in infancy or in later childhood. Thrombocytopenia with absent radii (TAR) is usually easily diagnosed in early infancy with a complete blood count and imaging studies demonstrating the presence of thumbs but absence of radii bilaterally. Thrombocytopenia usually improves as the child ages.
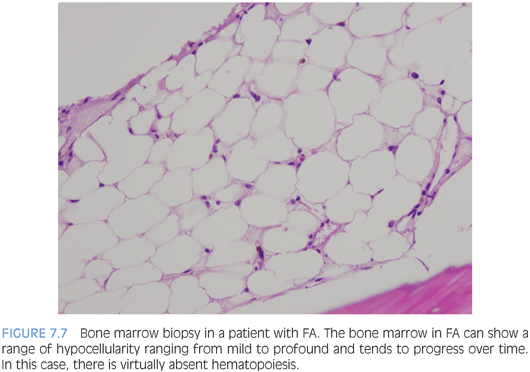
BONE MARROW FAILURE ASSOCIATED WITH MULTIPLE CYTOPENIAS. Inherited syndromes associated with multilineage bone marrow failure include a variety of disorders usually associated with other congenital abnormalities or clinical findings that may bring patients to medical attention, although it is now recognized that the clinical spectrum is broad and many patients may present initially with symptoms of marrow failure without other evident features of an inherited disorder.5 Although all of these disorders are rare, the relatively more common are Fanconi anemia (FA), dyskeratosis congenita (DKC), and SDS. Most patients will present sometime in childhood; however, patients with FA or DKC may present in early adulthood. Patients will present with one or more cytopenias of variable severity, and anemia, when present, is reflective of stress erythropoiesis, with macrocytosis and increased Hgb F. The bone marrow findings are similar for all patients and demonstrate a hypoplastic marrow of variable severity with a decrease in all three cell lines but normal maturation without dysplasia, unless the patient has evolved to a myelodysplastic syndrome (MDS) (Fig. 7.7). Therefore, the diagnosis rests on correlation with other clinical features, laboratory findings, and ancillary studies.
Acquired aplastic anemia accounts for the majority of patients who present with multilineage bone marrow failure and may present secondary to marrow insult due to drug or toxin exposure, infection, or after hepatitis, although most cases are thought to be immune mediated. Patients present with symptoms due to their cytopenias, pancytopenia, and a hypocellular marrow, which may be profoundly replaced by adipose tissue with little to no visible hematopoietic elements. Severity of aplastic anemia is graded as moderate, severe, or very severe, depending on the degree of the cytopenias and hypocellularity.6 Careful examination of residual hematopoietic elements for dysplastic features is important because many cases of low-grade pediatric MDS present with a hypocellular marrow (refractory cytopenia of childhood).
Histiocytic Disorders and Infiltrates
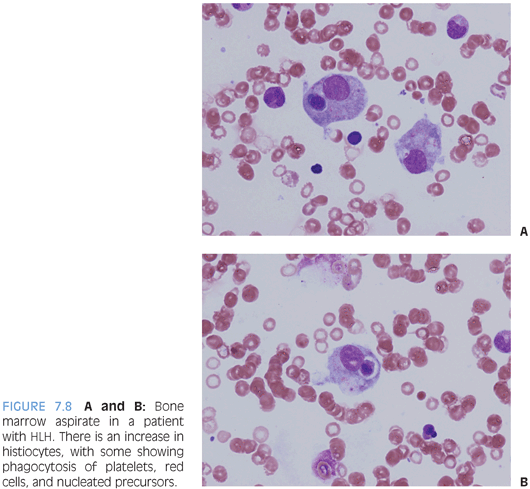
HEMOPHAGOCYTIC LYMPHOHISTIOCYTOSIS (HLH). HLH is a disorder of profound histiocyte proliferation and activation that arises either as a primary disorder due to a genetic mutation or secondary to an infection, lymphoma, or immunodeficiency, with Epstein-Barr virus (EBV) being an initiating trigger in many of these settings. Patients present with symptoms of cytokine storm with persistent fever, hepatosplenomegaly, cytopenias, hemophagocytosis in the bone marrow or another site, elevated triglycerides, and markers of inflammation including very high levels of ferritin and soluble interleukin-2 (IL-2). Along with a demonstrable mutation in the perforin gene, these features constitute the nine diagnostic criteria, of which five are necessary to establish the diagnosis.7 Patients require prompt treatment with steroids and chemotherapeutic agents to help get the process under control, as well as treatment of the underlying disorder, if it is a case of secondary HLH. Some patients will require bone marrow transplant, especially if it primary.
Bone marrow aspirate and biopsy will show increased lymphohistiocytic infiltrates with hemophagocytosis of erythrocytes and nucleated cells (Fig. 7.8). Erythrophagocytosis alone is not specific and may also be seen in the setting of recent transfusion. Sometimes, the infiltrates are subtle and the hemophagocytosis difficult to identify. Immunohistochemical studies for macrophage-specific markers such as the PGM1 clone of CD68 or CD163 can be helpful to highlight the infiltrates and hemophagocytosis. The background hematopoiesis is usually normal, and marrow cellularity may be normal or increased. Mature lymphocytes may be increased but appear normal in appearance unless the HLH is secondary to lymphoma with marrow involvement.
METABOLIC STORAGE DISEASES. Metabolic storage disorders are rare disorders that result from defects in an enzyme or other protein that interferes with a biochemical pathway and results in abnormal accumulation of a lipid, glycolipid, polysaccharide, or protein that cannot be broken down. The clinical spectrum is broad in severity, and the clinical manifestations depend on the precise defect. Because many of these products are ingested by macrophages in the reticuloendothelial system, the bone marrow frequently is involved by so-called storage disease histiocytes. Although peripheral blood manifestations with abnormal inclusions or vacuolization of white blood cells can be seen in many of these disorders, peripheral blood findings are uncommon, even in a patient with an established diagnosis.8
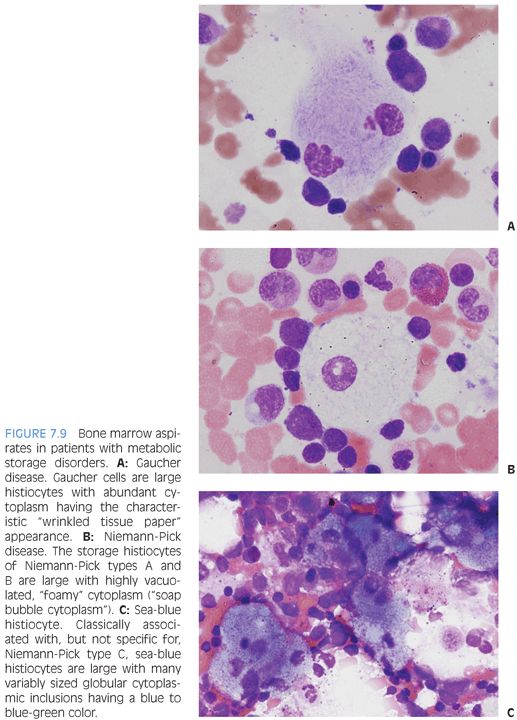
There are three main types of storage disease histiocytes (Gaucher cell, foamy macrophage, and sea-blue histiocyte), and the presence of these histiocytes, although characteristic, is not pathognomonic for a particular storage disease because many storage diseases may have a similar appearance in the bone marrow, and such histiocytes may be seen in a variety of clinical settings that are not related to storage disorders, such as the pseudo-Gaucher cells described in chronic myelogenous leukemia. Three of the prototypic diseases with their characteristic histiocyte are described in the following text.
Gaucher disease is due to a deficiency of glucocerebrosidase and is the most common of the lysosomal storage diseases. There are three main types, classified, in part, according to the presence or absence of neurologic involvement. Gaucher cells are large cells with abundant cytoplasm with linear inclusions giving the appearance of “crumpled tissue paper” or “wrinkled silk” (Fig. 7.9A). Pseudo-Gaucher cells can be seen in situations with highly increased cell turnover in the marrow, such as leukemias or defective erythropoiesis.
Niemann-Pick encompasses three main types (A, B, and C). Types A and B are due to acid sphingomyelinase deficiencies and result in hepatosplenomegaly with increased histiocytes in the reticuloendothelial system. The bone marrow shows “foam cells,” which are large histiocytes with abundant finely vacuolated or “soap bubble” cytoplasm (see Fig. 7.9B). Similar cells can be seen in patients with hereditary hyperlipidemias or other storage disorders. Niemann-Pick type C is due to defective cholesterol processing and transport and also results in hepatosplenomegaly. Classically, the bone marrow shows “sea-blue histiocytes,” which are macrophages with abundant blue-green to blue granular cytoplasmic inclusions (see Fig. 7.9C). A variety of other storage disorders will demonstrate similar “sea-blue histiocytes” in the bone marrow, as will clinical settings of increased cell turnover, such as acute or chronic leukemias. They have also been described in ITP.
NEOPLASMS
ACUTE LEUKEMIAS. Hematologic malignancies are more common than solid tumors in pediatrics, with B-acute lymphoblastic leukemia (B-ALL) being the most common childhood cancer overall. The very high cure rates achievable with current treatment regimens are the great success story of combination chemotherapy. Acute leukemias are classified by their morphology, lineage (immunophenotype), genetics, and clinical setting, following guidelines outlined by the WHO 2008 classification. It is recommended that pathology reports include all of these elements, making use of addenda to report cytogenetics and other molecular studies as they become available because the final classification is not complete without putting all of these elements together. In this way, the pathology report becomes “one-stop shopping” for all of the data the treating hematologist/oncologist requires to formulate a treatment plan.
ACUTE LYMPHOBLASTIC LEUKEMIA (ALL). ALL has a peak incidence between the ages of 2 and 5 years, and patients usually present acutely with bone pain and symptoms secondary to their cytopenias (bleeding/bruising, fever, fatigue, lethargy). They often have hepatosplenomegaly and may have peripheral lymphadenopathy. Patients with T-acute lymphoblastic leukemia (T-ALL) may have a mediastinal mass with pleural effusions. The complete blood count usually demonstrates anemia and thrombocytopenia, which may be severe, and a white blood cell count that may be normal, elevated, or low. Typically, the lactate dehydrogenase (LDH) is elevated, and uric acid may be elevated if the patient is in tumor lysis. Circulating blasts are usually seen on peripheral blood smear, but the number varies and, in some cases, may not be seen at all (the “aleukemic leukemia”). B-ALL is stratified by the WHO 2008 into several genetic subgroups, which are biologically and prognostically relevant, but T-ALL is not. Unlike acute myeloid leukemia, where the morphologic features may be predictive of the genetic underpinnings of the disease, for the most part, B-ALL or T-ALL has no such correlations for morphology.
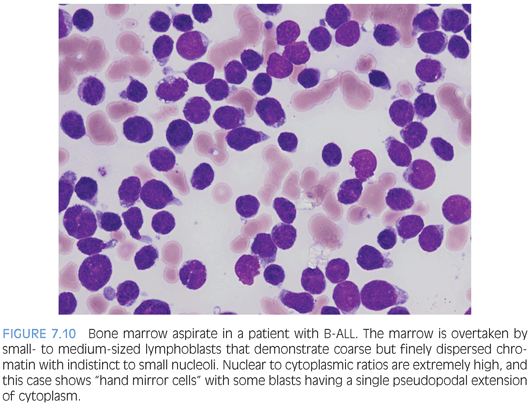
The morphology of lymphoblasts may vary and overlap with that of myeloblasts, highlighting the necessity of immunophenotyping for a definitive assignment of lineage. There are no distinct morphologic differences between B-ALL and T-ALL. In general, lymphoblasts are variable in size, but usually are small to intermediate and smaller than myeloblasts. The nuclear to cytoplasmic ratio is extremely high, with scant to more modest amounts of cytoplasm. The amount of cytoplasm may be so scant as to be barely visible but may be more prominent. Some cases may have one or more cytoplasmic vacuoles, and rarely, azurophilic granules may be seen. Auer rods are absent. The chromatin is evenly dispersed, delicately stippled, and powdery but, overall, coarser than myeloblasts with small to indistinct nucleoli. Some cases may have larger nucleoli and more open chromatin. Nuclear folds and clefts and irregular nuclear contours may be present (Fig. 7.10). On bone marrow biopsy, the marrow is usually extensively infiltrated (or at least >25% involved to distinguish between marrow involvement by lymphoblastic lymphoma) with little residual visible hematopoiesis remaining. Mitotic activity is usually robust (Fig. 7.11).