The Endocrinology of Pregnancy
 |
Who is in charge of pregnancy, the mother or her fetus? From the vantage point of an outsider looking in, it seems as if the mother is in charge. But from the fetal point of view, it is overwhelmingly logical that the maternal adaptations of pregnancy are controlled by the fetus. For the fetus, one of the crucial aspects of intrauterine life is its dependency on the effective exchange of nutritive and metabolic products with the mother. It is logical that mechanisms exist by which a growing fetus can influence or control the exchange process and, hence, its environment. The methods by which a fetus can influence its own growth and development involve a variety of messages transmitted, in many cases, by hormones. Hormonal messengers from the conceptus can affect metabolic processes, uteroplacental blood flow, and cellular differentiation. Furthermore, a fetus may signal its desire and readiness to leave the uterus by hormonal initiation of parturition. This chapter reviews the mechanisms by which the fetus establishes influence over important events during pregnancy. The important process of lactation is discussed in Chapter 16.
Steroid Hormones in Pregnancy
Steroidogenesis in the fetoplacental unit does not follow the conventional mechanisms of hormone production within a single organ. Instead, the final products result from critical interactions and interdependence of separate organ systems that individually do not possess the necessary enzymatic capabilities. It is helpful to view the process as consisting of a fetal compartment, a placental compartment (specifically the syncytiotrophoblast),
and a maternal compartment. Separately, the fetal and placental compartments lack certain steroidogenic activities. Together, however, they are complementary and form a complete unit that utilizes the maternal compartment as a source of basic building materials and as a resource for clearance of steroids.
and a maternal compartment. Separately, the fetal and placental compartments lack certain steroidogenic activities. Together, however, they are complementary and form a complete unit that utilizes the maternal compartment as a source of basic building materials and as a resource for clearance of steroids.
 |
Progesterone
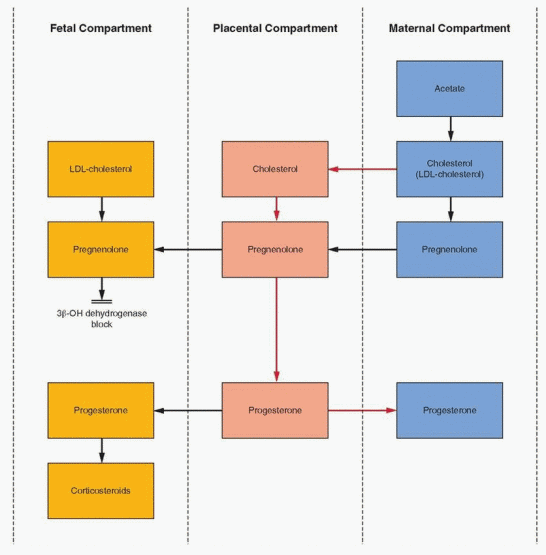
In its key location as a way station between mother and fetus, the placenta can use precursors from either mother or fetus to circumvent its own deficiencies in enzyme activity. The placenta converts little, if any, acetate to cholesterol or its precursors. Cholesterol and pregnenolone are obtained from the maternal bloodstream for progesterone synthesis. The fetal contribution is negligible because progesterone levels remain high after fetal demise. Thus, the massive amount of progesterone produced in pregnancy depends on placentalmaternal cooperation, although some have argued that the fetal liver is an important source of cholesterol (discussed later).
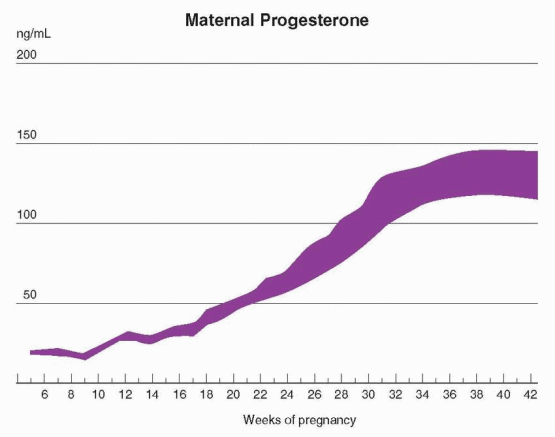
Progesterone is largely produced by the corpus luteum until about 10 weeks of gestation. Indeed, until approximately the seventh week, the pregnancy is dependent on the presence of the corpus luteum.1 Exogenous support for an early pregnancy (until 10 weeks) requires 100 mg of progesterone daily, associated with a maternal circulating level of approximately 10 ng/mL.2 Despite this requirement, patients pregnant after ovarian stimulation with one of the techniques of assisted reproductive technology have concluded a successful pregnancy after experiencing extremely low progesterone levels.3,4 Thus, individual variation is great, and very low circulating levels of progesterone can be encountered occasionally in women who experience normal pregnancies. The predictive value, therefore, of progesterone measurements is limited.
After a transition period of shared function between the seventh week and tenth week, during which there is a slight decline in circulating maternal progesterone levels, the placenta
emerges as the major source of progesterone synthesis, and maternal circulating levels progressively increase.2,5,6 At term, progesterone levels range from 100 to 200 ng/mL, and the placenta produces about 250 mg/day. Most of the progesterone produced in the placenta enters the maternal circulation.
emerges as the major source of progesterone synthesis, and maternal circulating levels progressively increase.2,5,6 At term, progesterone levels range from 100 to 200 ng/mL, and the placenta produces about 250 mg/day. Most of the progesterone produced in the placenta enters the maternal circulation.
In contrast to estrogen, progesterone production by the placenta is largely independent of the quantity of precursor available, the uteroplacental perfusion, fetal well being, or even the presence of a live fetus. This is because the fetus contributes essentially no precursor. The majority of placental progesterone is derived from maternal cholesterol that is readily available. At term a small portion (3%) is derived from maternal pregnenolone.
The cholesterol utilized for progesterone synthesis enters the trophoblast from the maternal bloodstream as low-density lipoprotein (LDL)-cholesterol, by means of the process of endocytosis (internalization, as described in Chapter 2) i nvolving the LDL cell membrane
receptors, a process enhanced in pregnancy by estrogen.7, 8 Hydrolysis of the protein component of LDL may yield amino acids for the fetus, and essential fatty acids may be derived from hydrolysis of the cholesteryl esters. Unlike steroidogenesis elsewhere, it is not clear whether placental progesterone production requires the control of tropic hormones. Although some evidence suggests tropic hormone support is not necessary, other evidence indicates that a small amount of human chorionic gonadotropin (hCG) must be present.9, 10
receptors, a process enhanced in pregnancy by estrogen.7, 8 Hydrolysis of the protein component of LDL may yield amino acids for the fetus, and essential fatty acids may be derived from hydrolysis of the cholesteryl esters. Unlike steroidogenesis elsewhere, it is not clear whether placental progesterone production requires the control of tropic hormones. Although some evidence suggests tropic hormone support is not necessary, other evidence indicates that a small amount of human chorionic gonadotropin (hCG) must be present.9, 10
 |
There is evidence in the baboon that estrogen (estradiol) regulates progesterone production in the placenta.11 The fetoplacental units in human and baboon pregnancies are virtually identical. Estradiol increases LDL-cholesterol uptake in baboon trophoblastic tissue by increasing LDL receptor gene transcription, and in human syncytiotrophoblast, estradiol increases progesterone production by means of an increase in LDL uptake.11, 12 Estrogen also stimulates cholesterol production in the human fetal liver to provide circulating LDL-cholesterol substrate for steroidogenesis.13 In addition, estrogen increases placental P450scc enzyme activity that converts cholesterol to pregnenolone, the immediate precursor for progesterone. Because estrogen production ultimately depends on the fetal adrenal gland for precursors, the influence of estrogen on progesterone production would be another example of fetal direction and control in the endocrinology of pregnancy. The proponents of this interaction and dependence of progesterone production on fetal precursors argue that the lack of impact by conditions of estrogen deficiency (e.g., anencephaly, fetal demise) on progesterone production is due to the fact that active, unbound estrogen remains within a critical, effective range, and what is lost reflects the degree of excess production in pregnancy.11
The human decidua and fetal membranes also synthesize and metabolize progesterone.14 In this case, neither cholesterol nor LDL-cholesterol are significant substrates; pregnenolone sulfate may be the most important precursor. This local steroidogenesis may play a role in regulating parturition.
Amniotic fluid progesterone concentration is maximal between 10 and 20 weeks and then decreases gradually. Myometrial levels are about 3 times higher than maternal plasma levels in early pregnancy, remain high, and are about equal to the maternal plasma concentration at term.
In early pregnancy, the maternal levels of 17a-hydroxyprogesterone rise, marking the activity of the corpus luteum. By the tenth week of gestation, this compound has returned to baseline levels, indicating that the placenta has little 17a-hydroxylase activity. However, beginning about the 32nd week there is a second, more gradual rise in 17ahydroxyprogesterone due to placental utilization of fetal precursors.
There are two active metabolites of progesterone that increase significantly during pregnancy. There is about a 10-fold increase of the 5a-reduced metabolite, 5a-pregnane3-20-dione.15 This compound contributes to the resistance in pregnancy against the vasopressor action of angiotensin II. The circulating level, however, is the same in normal and hypertensive pregnancies. The maternal blood concentration of deoxycorticosterone (DOC) at term is 1,200 times the nonpregnant levels. Some of this is due to the 3-4-fold increase in cortisol-binding globulin during pregnancy, but a significant amount is due to 21-hydroxylation of circulating progesterone in the kidney.16 This activity is significant during pregnancy because the rate is proportional to the circulating concentration of progesterone. The fetal kidney is also active in 21-hydroxylation of the progesterone secreted by the placenta into the fetal circulation. Currently, there is no known physiologic role for DOC during pregnancy.
Progesterone has a role in parturition as discussed later in this chapter. It has been suggested that progesterone is also important in suppressing the maternal immunologic response to fetal antigens, thereby preventing maternal rejection of the trophoblast. And, of course, progesterone prepares and maintains the endometrium to allow implantation. The human corpus luteum makes significant amounts of estradiol, but it is progesterone and
not estrogen that is required for successful implantation.17 Because implantation normally occurs about 5-6 days after ovulation, and human chorionic gonadotropin (hCG) must appear by the 10th day after ovulation to rescue the corpus luteum, the blastocyst must successfully implant and secrete hCG within a narrow window of time. In the first 5-6 weeks of pregnancy, hCG stimulation of the corpus luteum results in the daily secretion of about 25 mg of progesterone and 0.5 mg of estradiol. Although estrogen levels begin to increase at 4-5 weeks due to placental secretion, progesterone production by the placenta does not significantly increase until about 10-11 weeks after ovulation.
not estrogen that is required for successful implantation.17 Because implantation normally occurs about 5-6 days after ovulation, and human chorionic gonadotropin (hCG) must appear by the 10th day after ovulation to rescue the corpus luteum, the blastocyst must successfully implant and secrete hCG within a narrow window of time. In the first 5-6 weeks of pregnancy, hCG stimulation of the corpus luteum results in the daily secretion of about 25 mg of progesterone and 0.5 mg of estradiol. Although estrogen levels begin to increase at 4-5 weeks due to placental secretion, progesterone production by the placenta does not significantly increase until about 10-11 weeks after ovulation.
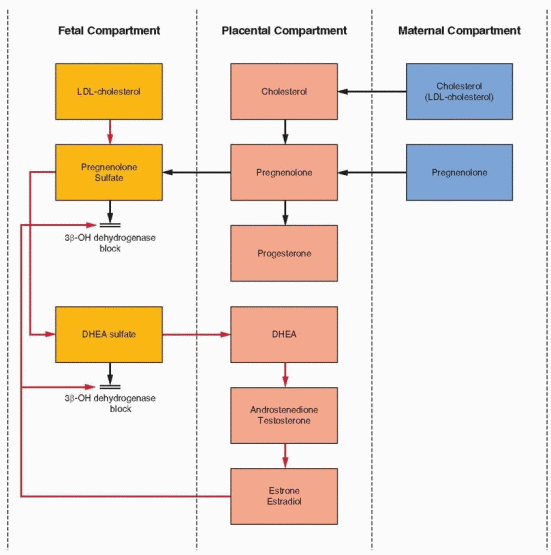
Progesterone serves as the substrate for fetal adrenal gland production of glucocorticoids and mineralocorticoids; however, cortisol synthesis is also derived from low-density lipoprotein cholesterol (LDL-cholesterol) synthesized in the fetal liver and obtained from the fetal circulation.13, 18 The fetal zone in the adrenal gland is extremely active but produces steroids with a 3β-hydroxy-Δ5 configuration like pregnenolone and dehydroepiandrosterone (DHEA), rather than 3-keto-Δ4 products such as progesterone. The fetus, therefore, lacks significant activity of the 3β-hydroxysteroid dehydrogenase, δ4 and 5 isomerase system. Thus, the fetus must borrow progesterone from the placenta to circumvent this lack in order to synthesize the biologically important corticosteroids. In return, the fetus supplies what the placenta lacks: 19-carbon compounds to serve as precursors for estrogens.
Steroid levels have been compared in maternal blood, fetal blood, and amniotic fluid obtained at fetoscopy in women undergoing termination of pregnancy at 16-20 weeks gestation.19 Cortisol, corticosterone, and aldosterone are definitely secreted by the fetal adrenal gland independently of the mother. The fetal arterial-venous differences confirm that placental progesterone is a source for fetal adrenal cortisol and aldosterone.
Estrogens
Estrogen production in pregnancy is under the control of the fetus and is a fundamental signaling method by which the fetus directs important physiologic processes that affect fetal well-being. Estrogen influences progesterone production, uteroplacental blood flow, mammary gland development, and fetal adrenal gland function.11
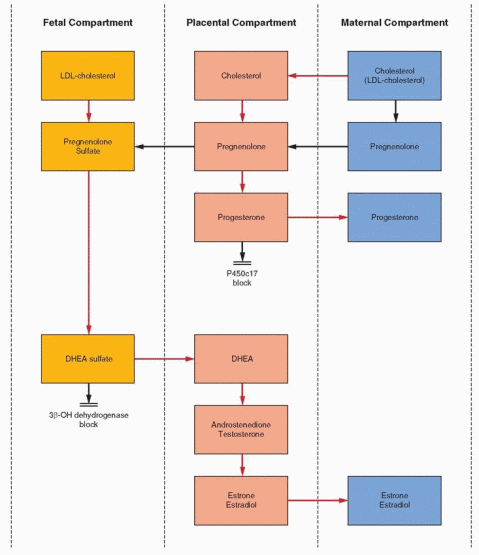
The basic precursors of estrogens are 19-carbon androgens. However, there is a virtual absence of 17a-hydroxylation and 17-20 desmolase (lyase) activity (P450c17) in the human placenta. As a result, 21-carbon products (progesterone and pregnenolone) cannot be converted to 19-carbon steroids (androstenedione and dehydroepiandrosterone). Like progesterone, estrogen produced by the placental aromatase (P450arom) enzyme system must derive precursors from outside the placenta.20
The androgen compounds utilized for estrogen synthesis in human pregnancy are, in the early months of gestation, derived from the maternal bloodstream. By the 20th week of pregnancy, the vast majority of estrogen excreted in the maternal urine is derived from fetal androgens. In particular, approximately 90% of estriol excretion can be accounted for by dehydroepiandrosterone sulfate (DHEAS) production by the fetal adrenal gland.20, 21 The high output of DHEAS by the fetal zone is due to low 3β-hydroxysteroid dehydrogenase gene expression.22 Removed into cell culture conditions, this gene becomes active in response to adrenocorticotropic hormone (ACTH).
The fetal endocrine compartment is characterized by rapid and extensive conjugation of steroids with sulfate. This is a protective mechanism, blocking the biologic effects of potent steroids present in such great quantities. In order to utilize fetal precursors, the placenta must be extremely efficient in cleaving the sulfate conjugates brought to it via the
fetal bloodstream. Indeed, the sulfatase activity in the placenta is rapid and quantitatively very significant. It is recognized that a deficiency in placental sulfatase is associated with low estrogen excretion, giving clinical importance to this metabolic step. This syndrome is discussed in greater detail later in this chapter.
fetal bloodstream. Indeed, the sulfatase activity in the placenta is rapid and quantitatively very significant. It is recognized that a deficiency in placental sulfatase is associated with low estrogen excretion, giving clinical importance to this metabolic step. This syndrome is discussed in greater detail later in this chapter.
 |
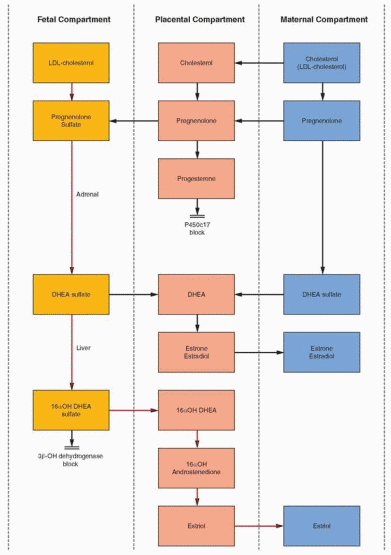
The fetal adrenal provides DHE AS as precursor for placental production of estrone and estradiol. However, the placenta lacks 16a-hydroxylation ability, and estriol with its 16a-hydroxyl group must be derived from an immediate fetal precursor. The fetal adrenal, with the aid of 16a-hydroxylation in the fetal liver, provides the 16a-hydroxydehydroepiandrosterone
sulfate for placental estriol formation. After birth, neonatal hepatic 16a-hydroxylation activity rapidly disappears. The maternal contribution of DHEAS to total estrogen synthesis must be negligible because, in the absence of normal fetal adrenal glands (as in an anencephalic infant), maternal estrogen levels and excretion are extremely low. The fetal adrenals secrete more than 200 mg of DHEAS daily, about 10 times more than the mother.23 Estriol is the estrogen produced in greatest quantity during pregnancy; estrone and estradiol are derived equally from fetal and maternal precursors.21 The profiles of the unconjugated compounds in the maternal compartment for the three major estrogens in pregnancy are as follows:
sulfate for placental estriol formation. After birth, neonatal hepatic 16a-hydroxylation activity rapidly disappears. The maternal contribution of DHEAS to total estrogen synthesis must be negligible because, in the absence of normal fetal adrenal glands (as in an anencephalic infant), maternal estrogen levels and excretion are extremely low. The fetal adrenals secrete more than 200 mg of DHEAS daily, about 10 times more than the mother.23 Estriol is the estrogen produced in greatest quantity during pregnancy; estrone and estradiol are derived equally from fetal and maternal precursors.21 The profiles of the unconjugated compounds in the maternal compartment for the three major estrogens in pregnancy are as follows:
 |
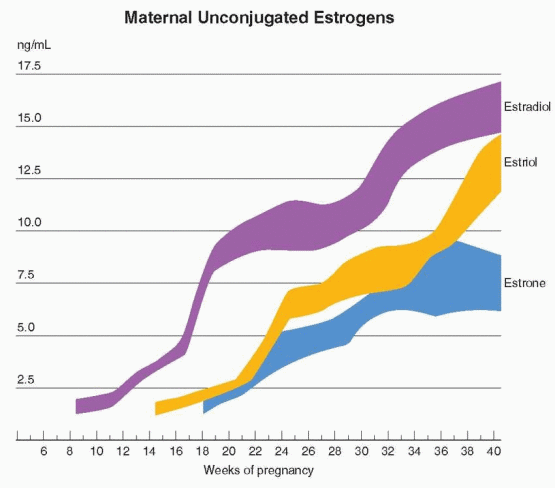 |
A rise in estrone begins at 6-10 weeks, and individual values range from 2 to 30 ng/mL at term.24 This wide range in normal values precludes the use of estrone measurements in clinical applications.
A rise in estradiol begins in weeks 6-8 when placental function becomes apparent.2 Individual estradiol values vary between 6 and 40 ng/mL at 36 weeks of gestation and then undergo an accelerated rate of increase.24 At term, an equal amount of estradiol arises from maternal DHEAS and fetal DHEAS, and its importance in fetal monitoring is negligible.
Estriol is first detectable at 9 weeks when the fetal adrenal gland secretion of precursor begins. Estriol concentrations plateau at 31-35 weeks and then increase again at 35-36 weeks.25
During pregnancy, estrone and estradiol production is increased about 100 times over nonpregnant levels. However, the increase in maternal estriol excretion is about a thousand-fold. The traditional view that estriol in pregnancy is a weak estrogen metabolite is not accurate. A weak estrogen provided in high concentrations can produce a biologic response
equivalent to that of estradiol.26 Because of its high production rate and concentration, estriol is an important hormone in pregnancy. The maternal level of estradiol is higher than in the fetus; in contrast, the estriol level in the fetus is greater than in the mother.
equivalent to that of estradiol.26 Because of its high production rate and concentration, estriol is an important hormone in pregnancy. The maternal level of estradiol is higher than in the fetus; in contrast, the estriol level in the fetus is greater than in the mother.
The maternal cardiovascular adaptations to pregnancy that are so necessary to serve the fetus are appropriately under the influence of the fetus and significantly regulated by estrogen.27 Blood volume is increased by estrogen stimulation of the maternal and trophoblastic renin-angiotensin systems, and uteroplacental blood flow, which is so critical for the fetus, is influenced by the vasodilatory effects of estrogen.
The enzyme responsible for estrogen synthesis is the cytochrome P450 aromatase enzyme (P450arom), the product of the CYP19 gene.28 The CYP19 gene is regulated in various tissues by tissue-specific promoters. The placenta, with its huge capacity for estrogen synthesis, uses a powerful, unique promoter that allows specific regulation. An autosomal-recessive disorder due to mutations in the P450arom gene is associated with a failure to convert androgen precursors to estrogen by placental aromatase.29 Consequently, a female fetus and the mother can undergo virilization. Nevertheless, growth and development of the fetus are not impaired, and this disorder raises the question: How much, if any, estrogen is essential in human pregnancy? Is this another example of backup mechanisms operating to achieve the goal?
Normally, placental aromatization is so efficient that little androgen presented to the placenta escapes.30 For this reason, fetuses are well protected against masculinization, and even in the presence of an androgen-secreting tumor, extremely large amounts of aromatizable androgens or the secretion of nonaromatizable androgens are required to produce unwanted virilization.
The estrogens presented to the maternal bloodstream are rapidly metabolized by the maternal liver prior to excretion into the maternal urine as a variety of more than 20 products. The bulk of these maternal urinary estrogens is composed of glucosiduronates conjugated at the 16-position. Significant amounts of the 3-glucosiduronate and the 3-sulfate-16-glucosiduronate are also excreted. Only approximately 8-10% of the maternal blood estriol is unconjugated.
The Fetal Adrenal Cortex
The fetal adrenal cortex is unique, differentiating by 8-9 weeks gestational age into a thick inner fetal zone and a thin outer definitive zone, which is the source of cortisol and the forerunner of the adult cortex.31 Early in pregnancy, adrenal growth and development are remarkable, and the gland achieves a size equal to or larger than that of the kidney by the end of the first trimester. After 20-24 weeks, the adrenal glands slowly decrease in size until a second spurt in growth begins at about 34-35 weeks. The gland remains proportionately larger than the adult adrenal glands. After delivery, the fetal zone (about 85% of the bulk of the gland) rapidly involutes to be replaced by simultaneous expansion of the outer definitive zone to form the zona glomerulosa, and the transitional zone to form the zona fasciculata and the zona reticularis (which expands again during adrenarche at puberty). By age 1, the fetal zone is gone, replaced by the adult adrenal cortex. Thus, the specific steroidogenic characteristics of the fetus are associated with a specific adrenal morphology that is dependent on specific factors present during intrauterine life.
Fetal dehydroepiandrosterone (DHEA) and DHEAS production rises steadily concomitant with the increase in the size of the fetal zone and adrenal weight.32 DHEA and DHEAS are the major secretory products of the fetal zone because 3β-hydroxysteroid dehydrogenase-isomerase activity and the expression of this enzyme’s gene are suppressed.22, 33 The well-known increase in maternal estrogen levels is significantly influenced by the increased availability of fetal DHEAS as a precursor. Indeed, the accelerated rise in maternal estrogen levels near term can be explained, in part, by an
increase in fetal DHEAS. The stimulus for the substantial adrenal growth and steroid production has been a puzzle.
increase in fetal DHEAS. The stimulus for the substantial adrenal growth and steroid production has been a puzzle.
Early in pregnancy, the adrenal gland can grow and function without ACTH, perhaps in response to hCG.31 After 15-20 weeks, fetal ACTH is required. However, during the last 12-14 weeks of pregnancy when fetal ACTH levels are declining, the adrenal quadruples in size.34 Because pituitary prolactin is the only fetal pituitary hormone to increase throughout pregnancy, paralleling fetal adrenal gland size changes, it was proposed that fetal prolactin is the critical tropic substance. In experimental preparations, however, only ACTH exerts a steroidogenic effect. There is no fetal adrenal response to prolactin, hCG, growth hormone, melanocyte-stimulating hormone (MSH), or thyrotropin-releasing hormone (TRH).35, 36 Furthermore, in patients treated with bromocriptine, fetal blood prolactin levels are suppressed, but DHEAS levels are unchanged.37 Nevertheless, interest in prolactin persists because both ACTH and prolactin can stimulate steroidogenesis in vivo in the fetal baboon.38
There is no question that, in the second half of pregnancy, ACTH is essential for the morphologic development and the steroidogenic mechanism of the fetal adrenal gland.39, 40 ACTH activates adenylate cyclase, leading to steroidogenesis. Soon the supply of cholesterol becomes rate limiting. Further ACTH action results in an increase in LDL receptors, leading to an increased uptake of circulating LDL-cholesterol, largely derived from the fetal liver.18 With internalization of LDL-cholesterol, hydrolysis by lysosomal enzymes of the cholesteryl ester makes cholesterol available for steroidogenesis. For this reason, fetal plasma levels of LDL are low, and after birth newborn levels of LDL rise as the fetal adrenal involutes. In the presence of low levels of LDL-cholesterol, the fetal adrenal is capable of synthesizing cholesterol de novo.41 Thus, near term, both de novo synthesis and utilization of circulating LDLcholesterol are necessary to sustain the high rates of DHEAS and estrogen formation. In addition, ACTH increases adrenal response by increasing the expression of its own receptor.42
The tropic support of the fetal adrenal gland by ACTH from the fetal pituitary is protected by placental estrogen. The placenta prevents cortisol that is present in higher levels in the mother from reaching the fetus by converting cortisol to cortisone. The converting enzyme, 11β-hydroxysteroid dehydrogenase, is abundantly expressed in syncytiotrophoblast at the interface between fetal tissue and maternal blood, and is stimulated by placental estrogen.43, 44 Regulation of this enzyme by estrogen thus influences fetal ACTH secretion. With increasing estrogen levels in late gestation, even greater 11β-hydroxysteroid dehydrogenase activity would result in even less maternal cortisol reaching the fetal circulation. Thus, it is proposed that near labor, fetal ACTH secretion increases, the fetal adrenal gland undergoes greater maturation, and fetal cortisol synthesis from endogenous cholesterol increases.45 A relative deficiency in 11β-hydroxysteroid dehydrogenase type 2 (the high affinity isoform) would expose the fetus to excessively high cortisol levels and is correlated with low birth weight, which in turn is correlated with insulin resistance, abnormal lipids, and hypertension in adult life.46,47 and 48 A reduction of type 2 11β-hydroxysteroid dehydrogenase activity has been reported in pregnancies complicated by smoking and preeclampsia, conditions known to be associated with intrauterine growth retardation.49,50 A similar reduction in activity has been documented with idiopathic intrauterine growth retardation, accompanied by a decreased ratio of cortisone to cortisol in umbilical artery blood.48
An interaction has been demonstrated in vitro between progesterone and the lipoxygenase pathways that lead to the products of arachidonic acid other than prostaglandins in regards to the regulation of 11β-hydroxysteroid dehydrogenase activity.51 Progesterone down-regulates 11β-hydroxysteroid dehydrogenase expression, as do the products of lipoxygenase activity. Because the lipoxygenase products increase progesterone output by trophoblast cells, an increase in lipoxygenase activity because of infection could increase progesterone levels, which in turn would decrease 11β-hydroxysteroid dehydrogenase activity, resulting in higher cortisol levels in the fetus with the consequences of stress and growth retardation.
It has been suggested that the increase in fetal cortisol secretion during normal pregnancy competes with progesterone for the glucocorticoid receptor in the placenta, thus blocking the inhibitory action of progesterone on corticotropin-releasing hormone (CRH) synthesis, leading to an increase in CRH.52 Placental production of CRH and the size of the fetal adrenal gland are closely correlated in several primates, both reaching a peak in humans at the time of parturition. The increase in CRH would augment fetal ACTH secretion, producing adrenal growth and even more fetal cortisol in a positive feedback relationship, as well as more DHEAS to serve as precursor for the increase in estrogen that occurs prior to parturition. However, fetal ACTH levels in the last half of pregnancy are not increasing, but slightly decreasing. It is significant that CRH, as demonstrated in vitro, also directly stimulates DHEAS synthesis by the fetal adrenal gland.53, 54
This is an important fetal placental-adrenal cycle. Cortisol from the adrenal gland increases placental CRH production; CRH induces ACTH receptor expression in the definitive zone of the fetal adrenal gland, leading to even greater adrenal cortisol secretion, and that in turn increases placental CRH biosynthesis as gestation advances.55 Direct stimulation of the fetal zone by CRH, supported by the presence of ACTH, augments the increasing production of DHEA and DHEAS required for estrogen synthesis in late gestation.54
Adrenal gland steroidogenesis involves autocrine and paracrine regulation by various growth factors.31 Fetal adrenal cells produce inhibin, and the alpha subunit (present only in inhibin) is preferentially increased by ACTH.56,57 In the fetal adrenal, the beta subunit is not expressed; thus, inhibin-A and activin-A are the principal forms.
Activin enhances ACTH-stimulated steroidogenesis while inhibiting mitogenesis in human fetal zone adrenal cells.57 This effect on steroidogenic activity is not present in adult adrenal cells. In vitro, activin enhances a shift in fetal adrenal cells from ACTH stimulation of DHEAS production to cortisol production. This shift is analogous to the shift that occurs after birth. Perhaps activin plays this role in the remodeling of the fetal zone in the newborn. A specific action for inhibin in fetal adrenal cells has not been described.
We should not expect the fetal adrenal gland to be an exception to the ubiquitous presence and actions of all growth factors.31 Basic fibroblast growth factor has potent mitogenic activity mediating the growth response of the fetal adrenal cortex to ACTH. Evidence indicates that the epidermal growth factor receptor is activated in the fetal adrenal, but the ligand using this receptor is probably transforming growth factor-a. Like activin, transforming growth factor-β inhibits fetal zone cellular proliferation and, in addition, suppresses steroidogenesis.
The insulin-like growth factors (IGF-I and IGF-II) are important in mediating the tropic effects of ACTH, particularly increasing adrenal responsiveness to ACTH in the second half of pregnancy.58 IGF-II production in the fetal adrenal is very significant and is stimulated by ACTH. IGF-II is believed to be important in prenatal growth.59 The abundance of IGF-II in the fetal adrenal gland implicates this growth factor as a mediator of ACTHinduced growth.60 Both IGF-I and IGF-II are equally mitogenic in a cell culture system of fetal adrenal cells and enhance the proliferation stimulated by basic fibroblast growth factor and epidermal growth factor.60 However, only transcription of IGF-II is stimulated by ACTH. IGF-II augments ACTH-stimulated steroidogenesis in the fetal adrenal, specifically by increasing the expression of P450c17.58 Thus, the growth-promoting and steroidogenic effects of ACTH are mediated by various growth factors, with a principal role played by IGF-II. In this regard, the fetal adrenal differs from the adult adrenal where IGF-I is predominant; however, IGF-II is able to modulate responsiveness to ACTH in the fetal adrenal by activating the IGF-I receptor.
Steroidogenic factor-1 (SF-1) and DAX-1 (named for the location of its gene on the X chromosome) are nuclear receptors for which specific ligands have not been identified (“orphan receptors”). SF-1 influences the expression of genes that encode steroidogenic enzymes, and when genetic expression of SF-1 is disrupted in mice, gonads and adrenal glands fail to develop.61,62
Mutations in the DAX-1 gene result in adrenal hypoplasia, and DAX-1 is believed to work with SF-1 in regulating development and function of steroid-producing tissues.63
Mutations in the DAX-1 gene result in adrenal hypoplasia, and DAX-1 is believed to work with SF-1 in regulating development and function of steroid-producing tissues.63
The production of DHEA is dependent on the CYP17 gene that is responsible for both 17ahydroxylase and 17,20-lyase enzyme activity. Differential regulation of these two activities with an increase in 17,20-lyase could account for the increase in DHEA in the fetal zone of the adrenal gland. The SULT2A1 gene is responsible for the sulfation and production of DHEAS. Modulation of this gene would also contribute to the steroidogenic activity of the fetal zone.
The unique features of the fetal adrenal gland can be ascribed to its high-estrogen environment. Tissue culture studies have demonstrated that hormonal peptides of pituitary or placental origin are not the factors that are responsible for the behavior of the fetal adrenal gland.64,65 and 66 Estrogens at high concentration inhibit 3β-hydroxysteroid dehydrogenase-isomerase activity in the fetal adrenal gland and, in the presence of ACTH in conjunction with IGF-II, enhance the secretion of DHEA.67 Estradiol concentrations of 10-100 ng/mL are required to inhibit cortisol secretion.68 The total estrogen concentrations in the fetus are easily in this range. A study of the kinetics of 3β-hydroxysteroid dehydrogenase activity in human adrenal microsomes reveals that all steroids are inhibitory, and most notably, estrone and
estradiol at levels found in fetal life cause almost total inhibition.69 In a study utilizing a human adrenocortical cell line, estradiol in high concentrations inhibited 3 β-hydroxy steroid dehydrogenase and the mechanism appeared to be independent of the estrogen receptor.70 The increase in DHEAS secretion by the fetal zone is a consequence of suppression of the gene (HSD3B2) that controls 3β-hydroxysteroid dehydrogenase expression; transcriptional factors necessary for the activity of this gene are absent in the fetal zone.71
estradiol at levels found in fetal life cause almost total inhibition.69 In a study utilizing a human adrenocortical cell line, estradiol in high concentrations inhibited 3 β-hydroxy steroid dehydrogenase and the mechanism appeared to be independent of the estrogen receptor.70 The increase in DHEAS secretion by the fetal zone is a consequence of suppression of the gene (HSD3B2) that controls 3β-hydroxysteroid dehydrogenase expression; transcriptional factors necessary for the activity of this gene are absent in the fetal zone.71
 |
The development of the adrenal gland during human fetal life and during the neonatal period is paralleled in the baboon.72 The adrenal cortex of the fetal baboon is characterized by the same deficiency in 3β-hydroxysteroid dehydrogenase as that seen in the human, with the same diversion of steroidogenesis into production of DHEA and DHEAS. Experimental studies in the baboon suggest that placental estrogen maintains the production of DHEA and DHEAS by the fetal adrenal cortex, but excessive adrenal growth and steroidogenesis is at the same time suppressed by the increasing estrogen levels in late pregnancy.73,74
Tissue growth in mammals is a consequence of cellular proliferation promoted by the cell regulators, cyclin D1 and cyclin E, which dimerize with kinases to form enzymes that carry out key phosphorylations during cell cycles. These key regulators are expressed in increasingly high concentrations in the baboon fetal adrenal cortex, beginning in early to mid gestation, especially in the outer definitive zone (destined to be the adult adrenal cortex and the source of cortisol).75 This early increase in the definitive zone is followed by a progressive decrease in these factors required for cellular proliferation in the definitive zone in late gestation. The cells in the fetal zone are derived from the definitive zone, and these changes indicate that the continued growth of the fetal adrenal cortex during gestation predominantly reflects cellular hypertrophy. Furthermore, because there is a progressive increase in the expression of 3β-hydroxysteroid dehydrogenase within the fetal definitive zone, the decline in proliferation is associated with functional differentiation as the definitive zone acquires the ability to produce mineralocorticoids and glucocorticoids.75 Here again, the key modulator of this change may be estrogen, specifically an estrogen-induced decrease in cyclin expression with advancing gestation.
The explanation that estrogen regulates 3β-hydroxysteroid dehydrogenase is challenged by in vitro studies of human fetal zone cells indicating that estradiol and IGF-II combine to direct steroidogenesis to DHEAS in a mechanism not due to inhibition of 3β-hydroxysteroid dehydrogenase.67 Nevertheless, it is an attractive and useful hypothesis to view the principal mission of the fetal adrenal as providing DHEAS as the basic precursor for placental estrogen production. Estrogen, in turn, feeds back to the adrenal to direct steroidogenesis along the Δ5 pathway to provide even more of its precursor, DHEAS. Thus far, this is the only known function for DHEAS. With birth and loss of exposure to estrogen, the fetal adrenal gland quickly changes to the adult type of gland. It seems reasonable to conclude that this complex change in the fetal adrenal cortex is orchestrated by the interplay among fetal pituitary ACTH, placental estrogen, and placental growth factors.
Measurement of Estrogen in Pregnancy
Because pregnancy is characterized by a great increase in maternal estrogen levels and estrogen production is dependent on fetal and placental steroidogenic cooperation, the amount of estrogen present in the maternal blood or urine reflects both fetal and placental enzymatic capability and, hence, well-being. Attention focused on estriol because 90% of maternal estriol is derived from fetal precursors. The end product to be assayed in the maternal blood or urine is influenced by a multitude of factors. Availability of precursor from the fetal adrenal gland is a prime requisite, as well as the ability of the placenta to perform its conversion steps. Maternal metabolism of the product and the efficiency of maternal renal excretion of the product can modify the daily amount of estrogen in the urine. Blood
flow to any of the key organs in the fetus, placenta, and mother becomes important.76,77 Fetal hypoxemia due to acute reductions in uteroplacental blood flow is associated with a marked increase in adrenal androgen production in response to an increase in fetal ACTH and, in response to the availability of androgen precursors, an increase in maternal estrogen levels.78 The response to acute stress is in contrast to the effect of chronic uteroplacental insufficiency, which is associated with a reduction in fetal androgens and maternal estrogens. In addition, drugs or diseases can affect any level in the cascade of events leading up to the assay of estrogen.
flow to any of the key organs in the fetus, placenta, and mother becomes important.76,77 Fetal hypoxemia due to acute reductions in uteroplacental blood flow is associated with a marked increase in adrenal androgen production in response to an increase in fetal ACTH and, in response to the availability of androgen precursors, an increase in maternal estrogen levels.78 The response to acute stress is in contrast to the effect of chronic uteroplacental insufficiency, which is associated with a reduction in fetal androgens and maternal estrogens. In addition, drugs or diseases can affect any level in the cascade of events leading up to the assay of estrogen.
For years, measurement of estrogen in a 24-hour urine collection was the standard hormonal method of assessing fetal well-being. This was replaced by immunoassay of unconjugated estriol in the plasma.79 Because of its short half-life (5-10 minutes) in the maternal circulation, unconjugated estriol has less variation than urinary or total blood estriol. However, assessment of maternal estriol levels has been superseded by various biophysical fetal monitoring techniques such as nonstress testing, stress testing, and measurement of fetal breathing and activity. Modern screening for fetal aneuploidy (discussed later in the chapter) utilizes three markers in the maternal circulation: alpha fetoprotein, human chorionic gonadotropin, and unconjugated estriol.
Amniotic Fluid Estrogen Measurements
Amniotic fluid estriol is correlated with the fetal estrogen pattern rather than the maternal. Most of the estriol in the amniotic fluid is present as 16-glucosiduronate or as 3-sulfate-16 glucosiduronate. A small amount exists as 3-sulfate. Very little unconjugated estriol is present in the amniotic fluid because free estriol is rapidly transferred across the placenta and membranes. Estriol sulfate is low in concentration because the placenta and fetal membranes hydrolyze the sulfated conjugates, and the free estriol is then passed out of the fluid. Because the membranes and the placenta have no glucuronidase activity, the glucosiduronate conjugates are removed slowly from the fetus. The glucosiduronates, therefore, predominate in the fetal urine and the amniotic fluid. Because of the slow changes in glucosiduronates, measurements of amniotic fluid estriol have wide variations in both normal and abnormal pregnancies. An important clinical use for amniotic fluid estrogen measurements has not emerged.
Estetrol
Estetrol (15α-hydroxyestriol) is formed from a fetal precursor and is very dependent on 15α-hydroxylation activity in the fetal liver. The capacity for 15a-hydroxylation of estrogens increases during fetal life, reaching a maximum at term. This activity then declines during infancy and is low, absent, or undetectable in adults. Estetrol may contribute to the estrogen effects taking place during pregnancy as maternal estetrol levels steadily increase with advancing gestation, and fetal levels are higher than maternal levels.80 Because of wide variations within and between individuals, there is no clinical use for maternal blood or urine estetrol measurements during pregnancy. However, estetrol, given in sufficient doses, is a potent, orally active estrogen that has potential for pharmacologic therapy.81
Placental Sulfatase Deficiency
There is an X-linked metabolic disease characterized by a placental sulfatase deficiency in the syncytiotrophoblast and postnatal ichthyosis, occurring in about 1 in 2,000-3,000
newborn males.82 Patients with the placental sulfatase disorder are unable to hydrolyze DHEAS or 16a-hydroxy-DHEAS; therefore, the placenta cannot form normal amounts of estrogen. A deficiency in placental sulfatase is usually discovered when patients go beyond term and are found to have extremely low estriol levels and no evidence of fetal distress. The patients usually fail to go into labor and require delivery by cesarean section. Most striking is the failure of cervical softening and dilation; thus, cervical dystocia occurs that is resistant to oxytocin stimulation. There are many case reports of this deficiency, almost all detected by finding low estriol levels. It was suggested that mothers who are carriers of this disorder are at increased risk for intrauterine growth retardation and perinatal complications even if the fetus is not affected.83 However, a careful analysis of unexplained low estriol levels concluded that this is a rare occurrence (about 3 per 10,000 pregnancies) and that perinatal complications in pregnancies at risk for placental sulfatase deficiency are not increased (other than a higher cesarean section rate).84 All newborn children, with a few exceptions, have been male. The steroid sulfatase X-linked recessive ichthyosis locus (the steroid sulfatase gene) has been mapped to the distal short arm portion of the X chromosome, Xp22.32.90% of cases of ichthyosis have a complete deletion of this gene plus flanking genes. There are no known geographic or racial factors that affect the gene frequency.
newborn males.82 Patients with the placental sulfatase disorder are unable to hydrolyze DHEAS or 16a-hydroxy-DHEAS; therefore, the placenta cannot form normal amounts of estrogen. A deficiency in placental sulfatase is usually discovered when patients go beyond term and are found to have extremely low estriol levels and no evidence of fetal distress. The patients usually fail to go into labor and require delivery by cesarean section. Most striking is the failure of cervical softening and dilation; thus, cervical dystocia occurs that is resistant to oxytocin stimulation. There are many case reports of this deficiency, almost all detected by finding low estriol levels. It was suggested that mothers who are carriers of this disorder are at increased risk for intrauterine growth retardation and perinatal complications even if the fetus is not affected.83 However, a careful analysis of unexplained low estriol levels concluded that this is a rare occurrence (about 3 per 10,000 pregnancies) and that perinatal complications in pregnancies at risk for placental sulfatase deficiency are not increased (other than a higher cesarean section rate).84 All newborn children, with a few exceptions, have been male. The steroid sulfatase X-linked recessive ichthyosis locus (the steroid sulfatase gene) has been mapped to the distal short arm portion of the X chromosome, Xp22.32.90% of cases of ichthyosis have a complete deletion of this gene plus flanking genes. There are no known geographic or racial factors that affect the gene frequency.
The characteristic steroid findings are as follows: extremely low estriol and estetrol levels in the mother with extremely high amniotic fluid DHEAS and normal amniotic fluid DHEA and androstenedione. The normal DHEA and androstenedione with a high DHEAS rule out congenital adrenal hyperplasia. The small amount of estriol that is present in these patients probably arises from 16a-hydroxylation of DHEAS in the maternal liver, thus providing 16a-hydroxylated DHEA to the placenta for aromatization to estriol. Maternal estrone and estradiol are also low but not as markedly reduced because of their utilization of maternal precursors. Measurement in maternal urine of steroids derived from fetal sulfated compounds is a simple and reliable means of prenatal diagnosis. Demonstration of a high level of DHEAS in the amniotic fluid is confirming. To establish the diagnosis with certainty, a decrease in sulfatase activity should be demonstrated in an in vitro incubation of placental tissue. The clinician should keep in mind that fresh tissue is needed for this procedure because freezing lowers enzyme activity. Alternatively, steroid sulfatase activity can be assayed in leukocytes.
It is now recognized that steroid sulfatase deficiency is present in other tissues and can persist after birth. These children develop ichthyosis beginning between birth and 6 months of age, characterized by hyperkeratosis (producing scales on the neck, trunk, and palms) and associated with mild corneal opacities, pyloric stenosis, and cryptorchidism. The skin fibroblasts have a low activity of steroid sulfatase, and scale formation that occurs early in the first year of life is thought to be due to an alteration in the cholesterol: cholesteryl ester ratio (due to the accumulation of cholesterol sulfate). This inherited disorder, thus, represents a single entity: placental sulfatase deficiency and X-linked ichthyosis, both reflecting a deficiency of microsomal sulfatase. More extensive deletions include contiguous genes and result in attention deficit hyperactivity disorder, autism, and mental retardation.85
A family history of scaling in males (as well as repeated postdate pregnancies and cesarean sections) should prompt a consideration for prenatal diagnosis. Because the clinical use of estriol measurements has declined, there is no effective method to identify the presence of this problem in women with normal obstetrical histories. However, a low maternal level of unconjugated estriol can be encountered with multiple marker screening (discussed later in this chapter). Furthermore, consideration should be given to antenatal screening by estriol measurement in pregnancies in which a male fetus is present and there is a previous history of a growth-retarded or stillborn male. However, perinatal outcome is good even when placental sulfatase deficiency is not known to be present and only a very small number of affected boys have serious manifestations of the disorder; therefore, it is difficult to justify the need for antenatal diagnosis.84
The Differential Diagnosis of an Extremely Low Estriol
Impending or present fetal demise.
Adrenal hypofunction.
Placental sulfatase deficiency.
Placental aromatase deficiency.
Drug-related effects.
Protein Hormones of Pregnancy
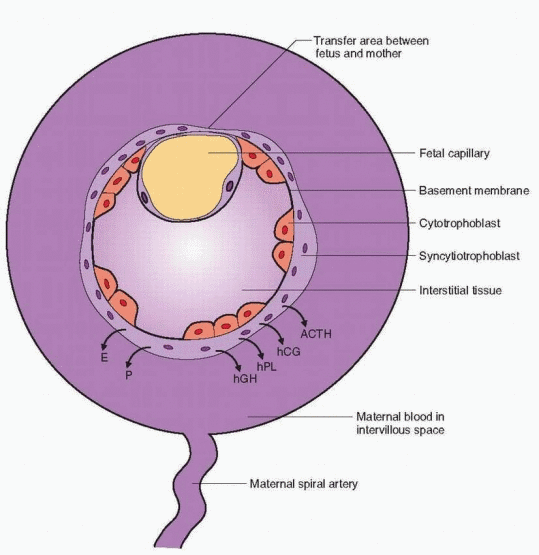
The placental villus is composed of trophoblast, mesenchymal cells, and fetal blood vessels. The two main trophoblastic layers consist of the cytotrophoblast, separate mononuclear cells prominent early in pregnancy and sparse late in pregnancy, and the syncytiotrophoblast, a continuous multinuclear layer on the surface of the villi. The cytotrophoblast is the basic placental stem cell from which the syncytiotrophoblasts arise by differentiation. The syncytiotrophoblast is, therefore, the functional cell of the placenta, the major site of hormone and protein production. Control of this important cellular differentiation is still not understood; however, the process is influenced by hCG and, undoubtedly, a variety of growth factors.86 The protein hormone system is complicated because individual peptides
can have multiple functions.87 The surface of the syncytiotrophoblast is in direct contact with the maternal blood in the intervillous space. This may be a reason why placental proteins are secreted preferentially into the mother.
can have multiple functions.87 The surface of the syncytiotrophoblast is in direct contact with the maternal blood in the intervillous space. This may be a reason why placental proteins are secreted preferentially into the mother.
 |
Proteins Associated with Pregnancy | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Hypothalamic-like Releasing Hormones
The human placenta contains many releasing and inhibiting hormones, including gonadotropin-releasing hormone (GnRH), corticotropin-releasing hormone (CRH), thyrotropin-releasing hormone (TRH) and somatostatin.88 Because of the presence of hypothalamic-like releasing hormones in an organ that produces tropic hormones, we are motivated to construct a system of regulation analogous to the hypothalamic-pituitary axis. However, as we shall see, this proves to be very difficult.
Immunoreactive GnRH can be localized in the cytotrophoblast and syncytiotrophoblast. Evidence indicates that placental GnRH regulates placental steroidogenesis and release of prostaglandins, as well as hCG.88,89,90,91 and 92 In some studies, the highest amount of GnRH was present early in pregnancy when the number of cytotrophoblasts is greatest and hCG secretion reaches its peak; however, others report relatively constant levels throughout pregnancy.93,94 All isoforms of GnRH are expressed in the human placenta, but GnRH-I is the predominant form.95,96
The placental receptors for GnRH have a lower affinity than that of GnRH receptors in the pituitary, ovary, and testis.97,98 This reflects the situation in which the binding site is in close proximity to the site of secretion for the regulatory hormone. A higher affinity is not necessary because of the large amount of GnRH available in the placenta, and the low-affinity receptors avoid response to the low levels of circulating GnRH. GnRH receptors, present in both cytotrophoblasts and syncytiotrophoblasts, are produced in a pattern that parallels the curve of hCG secretion, further evidence that placental GnRH and its receptor regulate hCG secretion.99 GnRH release is increased by estrogen, activin-A, insulin, and prostaglandins, and inhibited by progesterone, endogenous opiates, inhibin, and follistatin.87 The GnRH receptor is highly expressed in the fetal zone of the adrenal gland, raising the possibility of another pathway by which the placenta can influence fetal adrenal function.96
CRH, identical in structure to hypothalamic CRH, is produced in the trophoblast, the fetal membranes, and the decidua.87 Its production is regulated by steroids, decreased by progesterone, and, in contrast to the usual negative feedback action in the hypothalamus, increased by glucocorticoids.100 These interactions are consistent with the increase in fetal cortisol associated with the last weeks of pregnancy and the increase in ACTH with labor. Placental CRH is further regulated (as in the hypothalamus) by an array of substances such as vasopressin, norepinephrine, angiotensin II, prostaglandins, neuropeptide Y, and oxytocin. CRH release is stimulated by activin and interleukin and inhibited by inhibin and nitric oxide. The progressive increase in maternal CRH levels during pregnancy is due to the secretion of intrauterine CRH into the maternal circulation. The highest levels are found at labor and delivery. A binding protein for CRH exists in the human circulation, and it is produced in placenta, membranes, and decidua.101 Maternal levels of this binding protein are not different in pregnancy until a slight increase at 35 weeks, followed by a major decrease until term, increasing the bioavailability of CRH in late gestation. Maternal CRH levels are elevated in women with pregnancies under stress, e.g., with preeclampsia and preterm labor.87 An increase in placental CRH may be a response to the activation of fetal pituitary ACTH and adrenal cortisol secretion in the presence of hypoxemia. Placental CRH has multiple roles, including stimulation of the fetal adrenal plus parturition and regulation of blood flow. It is not certain to what extent CRH contributes to the increase in maternal adrenal secretion during pregnancy.
Trophoblast, amnion, chorion, and decidua also produce a peptide similar to CRH, named urocortin, that binds to CRH receptors and CRH-binding protein.102 Urocortin displays activities similar to CRH, such as inducing the secretion of prostaglandins and matrix metalloproteinases in placental cells and fetal membrane cells, and directly stimulating steroidogenesis in cells derived from the fetal zone of the adrenal gland.54,103,104
Human Chorionic Gonadotropin (hCG)
Human chorionic gonadotropin is a glycoprotein, a peptide framework to which carbohydrate side chains are attached.105 Alterations in the carbohydrate component (about one-third of the molecular weight) change the biologic properties. For example, the long half-life of hCG is approximately 24 hours as compared with 2 hours for luteinizing hormone (LH), a 12-fold difference, which is due mainly to the greater sialic acid content of hCG. As with the other glycoproteins, follicle-stimulating hormone (FSH), LH, and thyroid-stimulating hormone (TSH), hCG consists of two subunits, noncovalently linked by disulfide bonds, called alpha (α) and beta (β).106 The a subunit in these glycoprotein hormones is identical, consisting of 92 amino acids. Unique biologic activity as well as specificity in immunoassays is attributed to the molecular and carbohydrate differences in the β subunits (see “Heterogeneity” in Chapter 2).
β-hCG is the largest β subunit, containing a larger carbohydrate moiety and 145 amino acid residues, including a unique carboxyl terminal tailpiece of 24 amino acid groups. It is this unique part of the hCG structure that allows the production of highly specific antibodies and the utilization of highly specific immunologic assays. The extended sequence in the carboxyl-terminal region of β-hCG contains four sites for glycosylation, the reason why hCG is glycosylated to a greater extent than LH, a difference that is responsible for the longer circulating half-life for hCG.
All human tissues appear to make hCG, but the placenta is different in having the ability to glycosylate the protein, thus reducing its rate of metabolism and giving it biologic activity through a long half-life. The carbohydrate components of the glycoproteins are composed of fructose, galactose, mannose, galactosamine, glucosamine, and sialic acid. Although the other sugars are necessary for hormonal function, sialic acid is the critical determinant of biologic half-life. Removal of sialic acid residues in hCG, FSH, and LH leads to very rapid elimination from the circulation.
Genes for tropic hormones contain promoter and enhancer or inhibitor regions located in the 5′ flanking regions upstream from the transcription site. These sites respond to second messengers (cyclic AMP) as well as steroids and other yet unknown regulators. Differences in hCG structure are associated with a different promoter and transcriptional site that is located upstream in the hCG β subunit gene compared with the transcriptional site in the LH β subunit gene. The hCG β subunit promoter does not contain steroid hormone response elements, allowing hCG secretion to escape feedback regulation by the sex steroids, in contrast to FSH and LH.
The protein cores of the two glycoprotein subunits are the products of distinct genes.107 Using recombinant DNA technology, it has been demonstrated that there is a single human gene for the expression of the a subunit. The gene for the a subunit shared by FSH, LH, hCG, and TSH is located on chromosome 6p21.1-23. A single promoter site subject to multiple signals and hormones regulates transcription of the a-gene in both placenta and pituitary. The a subunit gene is expressed in several different cell types, but the β subunit genes are restricted in cell type. The TSH β-gene is expressed only in thyrotrophs regulated by thyroid hormone; the FSH β-gene is expressed in gonadotrophs regulated by GnRH, activin, inhibin, and gonadal steroids; the LH β-gene, also expressed in gonadotrophs, is regulated by GnRH and is unaffected by activin and inhibin.108
The α subunit gene requires the activation of distinct regulatory elements in thyrotroph and gonadotroph cells, as well as in the placenta. It is the activation of these cell-specific elements that produces tissue specificity for a-gene expression. In gonadotrophs, the GnRHsignaling pathway for a-gene transcription utilizes phosphorylase stimulation of diacylglycerol (DAG) and inositol triphosphate (IP3) that lead to a release of intracellular calcium
stores. GnRH also stimulates the influx of calcium at the cell membrane. DAG, IP3, and calcium work together to stimulate protein kinase C activity. Protein kinase regulation of the a-promoter is a principal part of the overall mechanism. This pituitary process is influenced by multiple factors including growth factors and gonadal steroids. In the placenta, the mechanism also utilizes specific regulatory elements, but the primary signal is mediated by the cyclic AMP-protein kinase A pathway.
stores. GnRH also stimulates the influx of calcium at the cell membrane. DAG, IP3, and calcium work together to stimulate protein kinase C activity. Protein kinase regulation of the a-promoter is a principal part of the overall mechanism. This pituitary process is influenced by multiple factors including growth factors and gonadal steroids. In the placenta, the mechanism also utilizes specific regulatory elements, but the primary signal is mediated by the cyclic AMP-protein kinase A pathway.
The genes that encode for the β subunits of LH, hCG, and TSH are located in a cluster on chromosome 19q13.3. There are six genes for the β subunit of hCG, and only one for β-LH.109 Transcription for the six hCG genes, each with different promoter activity, varies, and it is not certain why hCG requires multigenic expression (perhaps this is necessary to reach the extremely high level of production in early pregnancy). It is thought that β-hCG evolved relatively recently from β-LH, and the unique amino acid terminal extension of β-hCG arose by a read-through mutation of the translation stop codon in the β-LH gene; the DNA sequences of the β-hCG genes and the β-LH gene are 96% identical.109 Gene studies have indicated that the β-hCG gene originated in the common ancestor of monkeys, apes, and humans after the anthropoids diverged from tarsiers, about 35 to 55 million years ago.110,111
Only primates and equine species have been demonstrated to have genes for the β subunit of chorionic gonadotropin. In contrast to human chorionic gonadotropin, equine chorionic gonadotropin exerts both LH and FSH activities in many mammalian species because it contains peptide sequences in its β subunit that are homologous to those in the pituitary gonadotropins of other species. The equine β-chorionic gonadotropin gene is identical to the equine β-LH gene, and although the primate β-hCG gene evolved from the same ancestral β-LH gene, the equine chorionic gonadotropin gene evolved in a different way. The β-LH gene is not expressed in the placenta.
The genetic complexity for the transcription of β-hCG raises the possibility for mutations of these genes as causes of reproductive problems. A search for β-hCG gene deletions in women with recurrent miscarriage or unexplained infertility and for duplications in women with gestational trophoblastic neoplasia found only normal gene structures.112
hCG production and secretion are the result of complex interactions among the sex steroids, cytokines, GnRH, and growth factors. GnRH is synthesized by placental cells; GnRH receptors are present on placental cells; and GnRH stimulates the secretion of hCG and the steroid hormones in in vitro studies of placental cells.113,114 and 115 Similar responses can be demonstrated with other peptides, such as interleukin-1β.116 Similar to opiate action in the hypothalamus, the endorphins are a major inhibiting influence on hCG secretion.117 Also similar to the pituitary secretion of gonadotropins, inhibin restrains and activin enhances the GnRH-hCG system, with a positive influence of estrogen and a negative impact by progesterone.118,119 Follistatin, by binding activin, prevents the stimulatory activity of activin. Other growth factors, specifically IGF-I, IGF-II, TGF-a, and EGF, also influence hCG secretion.
Although a relatively clear story can be constructed into a working concept regarding the autocrine/paracrine interactions in the regulation of the menstrual cycle (Chapter 6), placental function is more complex, and a simple presentation of the many interactions cannot be produced. For example, epidermal growth factor stimulates hCG secretion, but also stimulates inhibin secretion in placental cells, and inhibin suppresses GnRH stimulation of hCG.120 Inhibin secretion in the placenta is further stimulated by prostaglandins.121
Can the cytotrophoblast-syncytiotrophoblast relationship be compared with the hypothalamic-pituitary axis? It does appear that hypothalamic-like peptides (CRH, GnRH) originate in the cytotrophoblast and influence the syncytiotrophoblast to secrete pituitary-like hormones (hCG, hPL, ACTH). Unraveling the interaction is made more difficult by the incredible complexity of the syncytiotrophoblast, a tissue that produces and responds to steroid and peptide hormones, growth factors, and neuropeptides. The best we can say is
that locally produced hormones, growth factors, and peptides work together to regulate placental function.
that locally produced hormones, growth factors, and peptides work together to regulate placental function.
To this day, the only definitely known function for hCG is support of the corpus luteum, taking over for LH on about the eighth day after ovulation, 1 day after implantation, when β-hCG first can be detected in maternal blood. hCG has been detected at the 8-cell stage in the embryo using molecular biology techniques.122 Continued survival of the corpus luteum is totally dependent on hCG, and, in turn, survival of the pregnancy is dependent on steroids from the corpus luteum until the seventh week of pregnancy.1 From the seventh week to the tenth week, the corpus luteum is gradually replaced by the placenta, and by the tenth week, removal of the corpus luteum will not be followed by steroid withdrawal abortion.
It is very probable, but not conclusively proven, that hCG stimulates steroidogenesis in the early fetal testes, so that androgen production will ensue, and masculine differentiation can be accomplished.123 However, normal masculine differentiation occurs in mouse models lacking LH receptors, and molecular evidence indicates that fetal Leydig cells (but not adult cells) respond to ACTH as well as hCG.124 It is also possible that the function of the inner fetal zone of the adrenal cortex depends on hCG for steroidogenesis early in pregnancy. The β-hCG gene is expressed in fetal kidney and fetal adrenal, suggesting that hCG may affect the development and function of these organs.125 In addition, hCG may regulate placental development by influencing the differentiation of cytotrophoblasts.126
hCG gene expression is present in both cytotrophoblast and syncytiotrophoblast, but it is synthesized mainly in the syncytiotrophoblast.127 The maternal circulating hCG concentration is approximately 100 IU/L at the time of the expected but missed menses. A maximal level of about 100,000 IU/L in the maternal circulation is reached at 8-10 weeks of gestation. Why does the corpus luteum involute at the time that hCG is reaching its highest levels? One possibility is that a specific inhibitory agent becomes active at this time. Another is down-regulation of receptors by the high levels of hCG. In early pregnancy, down-regulation may be avoided because hCG is secreted in an episodic fashion.128 For unknown reasons, the fetal testes escape desensitization; no receptor down-regulation takes place.123
hCG levels decrease to about 10,000-20,000 IU/L by 18-20 weeks and remain at that level to term. It is not certain why hCG levels are decreased in the second half of pregnancy. Advancing gestation is associated with increasing amounts of “nicked” hCG molecules in the maternal circulation.129 These molecules are missing a peptide linkage on the β-subunit, and, therefore, they dissociate into free α and β subunits. At any one point in time, the maternal circulation contains hCG, nicked hCG, free subunits, and fragments of hCG. In addition, the carbohydrate content of hCG varies throughout pregnancy, with more glycosylation present in early pregnancy (hyperglycosylated hCG). Overall, there are about 20-30 isoforms in the maternal blood, and the production of normal molecules is maximal in early gestation when the biologic actions of hCG are so important.130 A major route of clearance for hCG is renal metabolism in which a final reduced fragment of the β subunit is produced, known as the β-core fragment.
In the complex process of hCG regulation, several inhibiting factors have been identified, including inhibin and progesterone. The decline in hCG occurs at the time of increasing placental progesterone production, and a direct inhibition by this steroid could explain the lower levels of hCG after the tenth week of gestation.131
hCG levels close to term are higher in women bearing female fetuses. This is true of serum levels, placental content, urinary levels, and amniotic fluid concentrations. The mechanism and purpose of this difference are not known. Women who have markedly elevated levels of hCG in the second trimester, with no apparent explanation, have increased risks of spontaneous miscarriage, small-for-gestational-age infants, preeclampsia, and preterm delivery.132
There are two clinical conditions in which blood hCG titers are especially helpful: trophoblastic disease and ectopic pregnancies. Early pregnancy is characterized by the sequential appearance of hCG, followed by β-hCG and then a-hCG. The ratio of β-hCG to whole hCG remains constant after early pregnancy. Trophoblastic disease is distinguished by very high β-hCG levels (3-100 times higher than normal pregnancy). Ectopic production of a- and β-hCG by nontrophoblastic tumors is rare, but does occur.
In the United States, hydatidiform moles occur in approximately 1 in 600 induced abortions and 1 in 1,000-2,000 pregnancies. About 20% of patients with hydatidiform moles will develop malignant complications. Following molar pregnancies, the hCG titer should fall to a nondetectable level by 16 weeks in patients without persistent disease. Patients with trophoblastic disease show an abnormal curve (a titer greater than 500 IU/L) frequently by 3 weeks and usually by 6 weeks.133,134 A diagnosis of gestational trophoblastic disease is made when the β-HCG plateaus or rises over a 2-week period, or a continued elevation is present 16 weeks after evacuation. In the United States, the rare occurrence of this disease mandates consultation with a certified subspecialist in gynecologic oncology. Following treatment, hCG should be measured monthly for at least a year, then twice yearly for 5 years. To avoid missing the diagnosis of nonmolar trophoblastic disease, abnormal bleeding after any pregnancy should be evaluated with an hCG measurement, and all patients with elevated hCG levels and early pregnancy losses should be followed with serial hCG testing.
Choriocarcinoma is associated with the increased secretion of β-hCG that is glycosylated to a greater degree, so-called hyperglycosylated hCG, sometimes called invasive trophoblast antigen.135,136 Hyperglycosylated hCG detected in mothers in the first weeks of normal pregnancies is the major circulating form of hCG, but the levels decrease rapidly to be replaced by the usual hCG isoform by the second trimester.137 These findings suggest that hyperglycosylated hCG plays a role in regulating trophoblastic invasion; it is suggested that hyperglycosyalted hCG is mainly autocrine in its activity, whereas regular hCG functions as a classic hormone in maintaining the corpus luteum. Measurement of hyperglycosylated hCG in the first weeks of pregnancy may have a role in screening for Down syndrome, but clinical uses for assays that are specific for the many isoforms of hCG have yet to emerge.138, 139 Some of the inaccuracy associated with routine pregnancy testing, especially home pregnancy tests, can be attributed to the variability in detecting hyperglycosylated hCG.
We are just beginning to appreciate the complex heterogeneity of hCG, expressed by the many isoforms that are present in biological fluids.139 It is likely that a specific form of hCG can eventually be linked to a specific condition, offering the possibility of clinical application. For example, an assay specific for hyperglycosylated hCG may be of clinical value in assessing implantation and the early weeks of pregnancy; a low level predicts a failing pregnancy.140 General clinical use awaits improvements in available assays, requiring the development of pure standards and specific antibodies. Meanwhile, clinicians should keep in mind that current assays measure a pool of hCG and its various molecules.
Virtually all ectopic pregnancies are associated with detectable hCG. The hCG level increases at different rates in normal and ectopic pregnancies, and the quantitative measurement of hCG combined with pelvic ultrasonography has had an enormous impact on the diagnosis and management of ectopic pregnancy. This important clinical problem is discussed fully in Chapter 33. The contributions of hCG measurement can be summarized as follows:
The quantitative measurement of hCG can assess pregnancy viability. A normal rate of rise (at least a 50% increase every 2 days) usually indicates a normal pregnancy.
When the hCG titer exceeds 1,500-3,000 IU/L, vaginal ultrasonography should identify the presence of an intrauterine gestation.
Declining hCG levels are consistent with effective treatment, and persistent or rising levels indicate the presence of viable trophoblastic tissue.
With the use of modern sensitive assays, it is now appreciated that virtually all normal human tissues produce the intact hCG molecule. hCG can be detected in the blood of normal men and women, where it is secreted in a pulsatile fashion in parallel with LH; the source of this circulating hCG is the pituitary gland, perhaps a consequence of evolution when hCG was derived from LH.141,142,143 and 144 The concentration of this pituitary hCG normally approximates the sensitivity of the usual modern assay, and for this reason, many laboratories will not report the presence of hCG unless the level is 10 IU/L or higher. hCG produced in sites other than the placenta has little or no carbohydrate; therefore, it has a very short half-life and is rapidly cleared from the circulation. Significant levels of free a subunit are also present in the circulation of healthy individuals; however, the levels of the β subunit are extremely low.
False-positive Tests for hCG. False-positive results with the hCG assay occasionally occur and have been long-recognized, resulting in inappropriate surgical or medical treatment. The level is relatively low, usually less than 150 IU/L. There are many causes, including the hCG secreted by the pituitary, but this clinical problem is usually due to interference in the assay by other substances, especially antibodies to LH or anti-animal immunoglobulins.145 In addition, nontrophoblastic tumors can secrete detectable hCG. A false-positive result usually remains at the same level over time, neither increasing nor decreasing. When the clinical presentation is uncertain or not consistent with laboratory results (especially an absence of trophoblastic tissue), a positive hCG can be confirmed by several procedures:
Obtaining a similar result with a different assay method.
Demonstrating the presence of hCG in the urine.
Demonstrating parallel results with serial dilutions of the hCG standard and the patient’s serum sample.
Human Placental Lactogen (hPL)
Human placental lactogen (sometimes called human chorionic somatomammotropin), also secreted by the syncytiotrophoblast, is a single-chain polypeptide of 191 amino acids held together by 2 disulfide bonds. It is very similar in structure to human growth hormone (hGH), but has only 3% of hGH somatotropin activity. The growth hormonehPL gene family consists of 5 genes on chromosome 17q22-q24. Two genes encode for hGH, one in the pituitary and one in the placenta, and three for hPL; however, only two of the hPL genes are abundantly active in the placenta, each producing the same hPL hormone.146 The third hPL gene does generate a protein in the placenta, but its activity is limited.147
The half-life of hPL is short, about 15 minutes; hence its appeal as an index of placental problems. The level of hPL in the maternal circulation is correlated with fetal and placental weight, steadily increasing until plateauing in the last 4 weeks of pregnancy (5-10 μg/mL). There is no circadian variation, and only minute amounts of hPL enter the fetal circulation. Very high maternal levels are found in association with multiple gestations; levels up to 40 μ/mL have been found with quadruplets and quintuplets. An abnormally low level is anything less than 4 μg/mL in the last trimester.
 |
Physiologic Function
Although hPL is similar in structure to growth hormone, neither growth hormone-releasing hormone nor somatostatin influence placental hPL secretion. One would expect the regulatory mechanism to involve placental growth factors and cytokines, as is the case with other placental steroids and peptides. Although hPL has about 50% of the lactogenic activity of sheep prolactin in certain bioassays, its lactogenic contribution in human pregnancy is uncertain.
In the mother, hPL stimulates insulin secretion and IGF-I production and induces insulin resistance and carbohydrate intolerance. However, the well-recognized insulin resistance in pregnancy is not solely an effect of hPL; for example, placental cytokines (especially TNF-α) influence this metabolic state.148 Experimentally, the maternal level of hPL can be altered by changing the circulating level (chronically, not acutely) of glucose. hPL is elevated with hypoglycemia and depressed with hyperglycemia. This response in placental hPL may be secondary to the glucose-mediated changes in insulin levels; in vitro experiments with placental tissue indicated a decrease in hPL with a decrease in glucose, followed by an increase
in hPL after exposure to insulin.149 This information and studies in fasted pregnant women have led to the following formulation for the physiologic function of hPL.150,151,152,153,154,155 and 156
in hPL after exposure to insulin.149 This information and studies in fasted pregnant women have led to the following formulation for the physiologic function of hPL.150,151,152,153,154,155 and 156
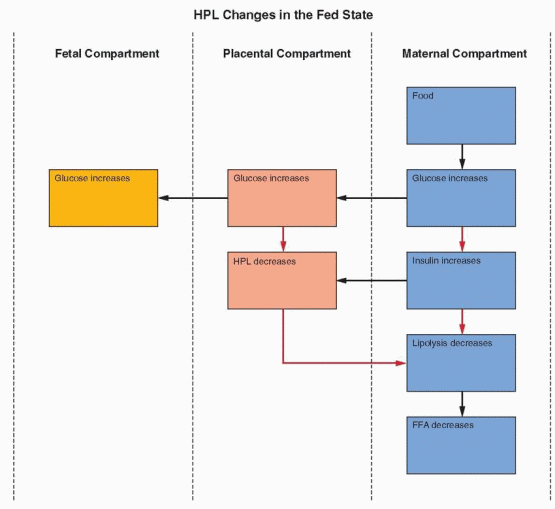 |
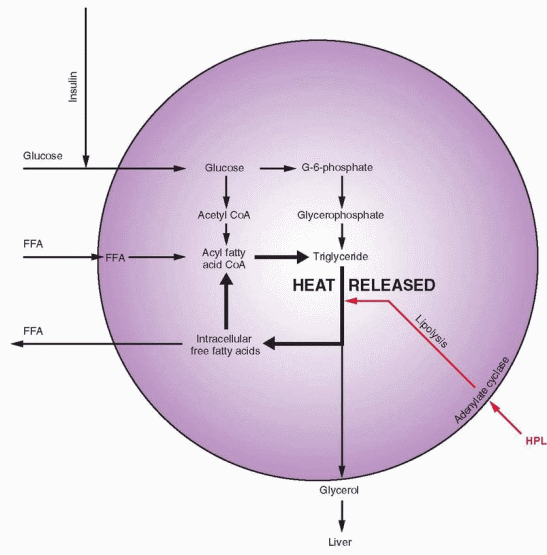
The metabolic role of hPL is to mobilize lipids as free fatty acids. In the fed state, there is abundant glucose available, leading to increased insulin levels, lipogenesis, and glucose utilization. This is associated with decreased gluconeogenesis and a decrease in the circulating free fatty acid levels, because the free fatty acids are utilized in the process of lipogenesis to deposit storage packets of triglycerides (see Chapter 19, Obesity).
Pregnancy has been likened to a state of “accelerated starvation,” characterized by a relative hypoglycemia in the fasting state.153 This state is due to two major influences:
Glucose provides the major, although not the entire, fuel requirement for the fetus. A difference in gradient causes a constant transfer of glucose from the mother to the fetus.
Placental hormones, specifically estrogen and progesterone, and especially hPL, interfere with the action of maternal insulin. In the second half of pregnancy when
hPL levels rise approximately 10-fold, hPL is a major force in the diabetogenic effects of pregnancy. The latter is characterized by increased levels of insulin associated with decreased cellular response (hyperinsulinemia and peripheral insulin resistance).
As glucose decreases in the fasting state, hPL levels rise. This stimulates lipolysis leading to an increase in circulating free fatty acids. Thus, a different fuel is provided for the mother so that glucose and amino acids can be conserved for the fetus. With sustained fasting, maternal fat is utilized for fuel to such an extent that maternal ketone levels rise. There is limited transport of free fatty acids across the placenta. Therefore, when glucose becomes scarce for the fetus, fetal tissues utilize the ketones that do cross the placenta. Thus, decreased glucose levels lead to decreased insulin and increased hPL, increasing lipolysis and ketone levels. hPL also may enhance the fetal uptake of ketones and amino acids. The mechanism for the insulin antagonism by hPL may be the hPL-stimulated increase in free fatty acid levels, which, in turn, directly interfere with insulin-directed entry of glucose into cells. These interactions significantly involve growth factors, particularly insulin-like growth factor, at the cellular level.
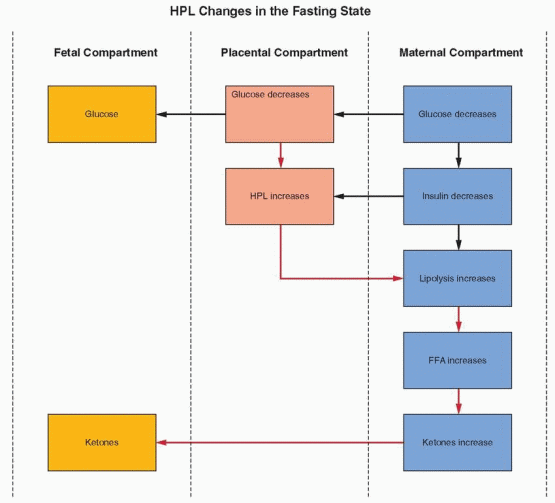 |
This mechanism can be viewed as an important means to provide fuel for the fetus between maternal meals. However, with a sustained state of inadequate glucose intake, the subsequent ketosis may impair fetal brain development and function. Pregnancy is not the time to severely restrict caloric intake. Indeed, impaired fetal growth and development are now recognized to correlate with adverse cardiovascular risk factors and disease in adult life as well as diabetes mellitus.47,157,158
The lipid, lipoprotein, and apolipoprotein changes during pregnancy are positively correlated with changes in estradiol, progesterone, and hPL.159 The lipolytic activity of hPL is an important factor because hPL is also linked to the maternal blood levels of cholesterol, triglycerides, phospholipids, and insulin-like growth factor-I.
When glucose is abundant, as in pregnant women with diabetes mellitus, the flow of nutritional substrates (in this case, glucose and amino acids) is in the direction of the fetus. The subsequent hyperinsulinemia in the fetus becomes a strong stimulus to growth, perhaps compounded by maternal hyperinsulinemia caused by obesity as well as the hyperinsulinemia due to the peripheral resistance produced by the hormones of pregnancy.160 Fetal undernutrition lowers fetal IGF-I levels, and this is associated with a high prevalence of insulin resistance later as adults.161 In vitro studies indicate that hPL, despite its lower levels in the fetus, directly affects fetal tissue metabolism, including synergistic actions with insulin, especially on glycogen synthesis in the liver. The failure of fetal growth hormone to affect fetal growth (e.g., normal growth in anencephalics) further indicates that hPL may be the fetal growth hormone.
hPL Clinical Uses
Blood levels of hPL are related to placental function. Some studies indicated that hPL was valuable in screening patients for potential fetal complications, but others did not support the use of hPL measurements. Although use of the hPL assay can have an impact on perinatal care, fetal heart rate-monitoring techniques are more reliably predictive and sensitive for assessing fetal well-being. Furthermore, totally uneventful pregnancies have been reported, despite undetectable hPL.162,163
Previous suggestions that a low or declining level of hPL and a high level of hCG are characteristic of trophoblastic disease were not accurate. Because of the rapid clearance of hPL (half-life of about 20 minutes), aborting molar pregnancies are likely to have low levels of hPL, whereas the level of hCG is still high. However, intact molar pregnancies can have elevated levels of both hPL and hCG.164
Human Chorionic Thyrotropin (hCT)
The human placenta contains two thyrotropic substances. One is called human chorionic thyrotropin (hCT), which is similar in size and action to pituitary TSH. The content in the normal placenta is very small, and it is unlikely that it has any physiologic importance. hCT differs from the other glycoproteins in that it does not appear to share the common a subunit. Antiserum generated to a-hCG does not neutralize the biologic activities of hCT, but it does neutralize that of hCG and pituitary TSH.
Rarely, patients with trophoblastic disease have hyperthyroidism, and even more rarely, thyroid storm.165 hCG has intrinsic thyrotropic activity, indicating that hCG is the second placental thyrotropic substance.166,167 and 168 It has been calculated that hCG contains approximately
1/4,000th of the thyrotropic activity of human TSH. In conditions with very elevated hCG levels, such as hydatidiform mole, the thyrotropic activity can be sufficient to produce hyperthyroidism (with elevated free thyroxine, FT4, but suppressed levels of thyroid-stimulating hormone, TSH), and this can even be encountered in normal pregnancy.169 Another very rare cause of hyperthyroidism in pregnancy is an inherited mutation of the thyrotropin receptor that makes it hypersensitive to hCG.170
1/4,000th of the thyrotropic activity of human TSH. In conditions with very elevated hCG levels, such as hydatidiform mole, the thyrotropic activity can be sufficient to produce hyperthyroidism (with elevated free thyroxine, FT4, but suppressed levels of thyroid-stimulating hormone, TSH), and this can even be encountered in normal pregnancy.169 Another very rare cause of hyperthyroidism in pregnancy is an inherited mutation of the thyrotropin receptor that makes it hypersensitive to hCG.170
There is a correlation between elevated thyroid function and hyperemesis gravidarum.171, 172 Hyperemesis gravidarum is usually associated with very high hCG levels, and some of these patients develop hyperthyroidism as well.173 Although free T4 will be elevated and TSH suppressed, patients with gestational hyperthyroidism do not develop the clinical signs of Graves’ disease and TSH-receptor antibodies, TRAb, will not be detectable. These hyperthyroid manifestations in normal pregnancies may be linked to a specific subpopulation of hCG molecules with greater thyrotropic bioactivity (because highly purified, standard hCG has only trivial TSH-like activity).174 Specifically, hCG with reduced sialic acid content is increased in pregnant patients with hyperemesis and hyperthyroidism.175 The thyroid hormone changes in pregnancy and the role of hCG as a thyroid stimulator are also discussed in Chapter 20.
Human Chorionic Adrenocorticotropin
The rise in maternal free cortisol and aldosterone that takes place throughout pregnancy is due to placental ACTH and corticotropin-releasing hormone (CRH) production and secretion into the maternal circulation and due to the effects of estrogen and progesterone on the maternal hypothalamic-pituitary axis.176,177 and 178 The placental content of ACTH is higher than can be accounted for by the contribution of sequestered blood. In addition, cortisol levels in pregnant women are resistant to dexamethasone suppression, indicating that there is a component of maternal ACTH and CRH that does not originate in the maternal hypothalamus and pituitary gland. The placental production of ACTH in the syncytiotrophoblast (and the increase in maternal ACTH levels) is probably due to stimulation by the locally produced CRH in the cytotrophoblast.179 Placental pro-opiomelanocortin (POMC) gene expression and ACTH content are present throughout pregnancy and increase in the weeks before term.180 One can speculate that placental ACTH and CRH raise maternal adrenal activity in order to provide the basic building blocks (cholesterol and pregnenolone) for placental steroidogenesis. The increased activity of the maternal adrenal gland is also necessary for the expansion of maternal blood volume during pregnancy.
The maternal ACTH response to the administration of CRH during pregnancy is blunted, reflecting a high level of endogenous CRH and ACTH activity. Vasopressin stimulates ACTH secretion in the pituitary, both directly and indirectly by potentiating the action of CRH. In contrast to the blunted response to CRH during pregnancy, the ACTH response to vasopressin is increased.181 This is further evidence that placental CRH produces a state of chronic stimulation for the maternal pituitary-adrenal axis. Thus, in contrast to nonpregnant women, CRH levels in maternal plasma are relatively high, rising in the second trimester to peak values at term.182,183 In contrast to the hypothalamic-pituitary axis, placental CRH and ACTH are not suppressed by glucocorticoids, and, therefore, maternal ACTH levels are little affected by corticosteroid administration to the mother. Oxytocin is a potent stimulator of CRH and ACTH placental production, a logical mechanism to meet the stress of labor and delivery. Binding of CRH with the CRH-binding protein blunts physiologic response, but the binding protein capacity is reached late in pregnancy, increasing the biological activity of CRH and further increasing cortisol availability during labor and delivery.184
Both maternal and fetal levels of CRH are further elevated in pathologic states such as premature labor, hypertension, fetal asphyxia, and intrauterine growth retardation.185 Because
CRH also stimulates prostaglandin synthesis in the placenta and fetal membranes, it is implicated in the premature labor that accompanies pathologic conditions.186
CRH also stimulates prostaglandin synthesis in the placenta and fetal membranes, it is implicated in the premature labor that accompanies pathologic conditions.186
Growth Hormone, Growth Hormone-Releasing Hormone, and Somatostatin
One of the two growth hormone genes on chromosome 17 is expressed only in the syncytiotrophoblast of the placenta, the other is expressed in the pituitary.147,187 The placental growth hormone is not identical to pituitary growth hormone, differing in 13 amino acids, and after 15-20 weeks of pregnancy, placental growth hormone gradually replaces pituitary growth hormone in the maternal circulation.147,188 Indeed, by term, maternal pituitary growth hormone is undetectable. Placental growth hormone is not present in fetal blood. The changes in maternal levels of insulin-like growth factors and insulin-like growth factor binding proteins reflect regulation by this placental growth hormone.189 Maternal IGF-I levels in the circulation increase during pregnancy in a pattern similar to that of placental growth hormone. Placental growth hormone is not regulated by placental growth hormone-releasing hormone but responds inversely to maternal glucose and insulin levels, protecting glucose availability for the fetus.147,190 Placental growth hormone can also stimulate gluconeogenesis and lipolysis in maternal organs. It is believed, therefore, that placental growth hormone influences fetal growth by affecting maternal metabolism. Placental growth hormone and maternal IGF-1 levels are lower in pregnancies with intrauterine growth retardation and higher in women with female fetuses.189,191 Maternal circulating levels of placental growth hormone are higher at midgestation in pregnancies with fetal Down syndrome.192
Alpha-Fetoprotein
Alpha-fetoprotein (AFP) is a relatively unique glycoprotein (590 amino acids and 4% carbohydrate) derived largely from fetal liver and partially from the yolk sac until it degenerates at about 12 weeks. In early pregnancy (5-12 weeks), amniotic fluid AFP is mainly from yolk sac origin, whereas maternal circulating AFP is mainly from the fetal liver.193 Its function is unknown, but it is comparable in size to albumin and contains 39% sequence homology; it may serve as a protein carrier of steroid hormones in fetal blood. AFP may also be a modulator of cell proliferation, synergizing with various growth factors.194
Peak levels of AFP in the fetal blood are reached at the end of the first trimester; then levels decrease gradually until a rapid decrease begins at 32 weeks. Maternal blood levels are much lower than fetal levels, rising until week 32 (probably because of the great increase in trophoblast villous surface area during this time period) and then declining. Because AFP is highly concentrated in the fetal central nervous system, abnormal direct contact of CNS with the amniotic fluid (as with neural tube and abdominal wall defects) results in elevated amniotic fluid and maternal blood levels. Other fetal abnormalities, such as intestinal obstruction, omphalocele, and congenital nephrosis, are also associated with high levels of AFP in the amniotic fluid. Besides indicating a variety of fetal anomalies, elevated maternal AFP levels are also present with multiple pregnancies and associated with an increased risk of spontaneous miscarriage, stillbirth, preterm birth, preeclampsia, neonatal death, and low birth weight (probably reflecting an increase in villous surface area in response to an adverse intrauterine environment).195,196 and 197 Conversely, very low maternal AFP levels are associated with large birth weight infants, miscarriage, and stillbirth.197,198
Multiple Marker Screening
Down syndrome is a very common genetic cause of abnormal development. The majority of cases are due to trisomy 21, an extra chromosome usually due to nondisjunction in maternal meiosis. A low maternal level of AFP is associated with trisomy 21. However, there is extensive overlap between normal and affected pregnancies responsible for a significant false-positive rate. Several placental products are secreted in increased amounts in pregnancies with trisomy 21, including hCG and hPL, whereas the maternal circulating level of unconjugated estriol is lower in affected pregnancies. The free β subunit of hCG usually circulates in low concentrations, but in the presence of a fetus with Down syndrome, the levels are high. With trisomy 18, all markers are decreased. Modern screening for fetal aneuploidy combines three markers: AFP, β-hCG, and unconjugated estriol.199,200 and 201 This protocol will detect 85% of open neural tube defects and 80% of Down syndrome, if gestational age is determined by ultrasonography.202 However, Down syndrome represents only about 50% of the chromosomal abnormalities that can be detected.
The multiple marker screening protocol measures AFP, unconjugated estriol, and hCG in maternal serum at 16-18 weeks gestation, the optimal time for neural tube defect detection. Using the patient’s age and the laboratory results, patients are provided a statistical estimation of risks for both neural tube defects and Down syndrome. Corrections are applied for race and weight. A pattern similar to that of Down syndrome has also been reported to be associated with hydropic fetal Turner syndrome.203
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


