Amenorrhea
 |
Few problems in gynecologic endocrinology can present a diagnostic challenge to clinicians like that of amenorrhea. The number, variety, and complexity of diseases and disorders that must be considered can seem daunting and, in many instances, includes unfamiliar organ systems. Moreover, some of the diagnostic possibilities can have serious consequences, if not recognized and treated effectively. Consequently, otherwise confident and experienced clinicians may view the problem as too complicated and time-consuming or may question their ability to perform or interpret the evaluation. However, when approached logically and systematically, the diagnostic evaluation of amenorrhea truly is straightforward, involving laboratory tests and procedures already familiar to almost all clinicians. With few exceptions, an evaluation can be completed quickly and without great expense.
The purpose of this chapter is to provide a systematic strategy for the evaluation of amenorrhea that will yield an accurate diagnosis, no matter how common or uncommon the cause. Once a diagnosis is established, additional corroborating evidence and the assistance of appropriate specialists (e.g., neurosurgeon, internist, endocrinologist, or psychiatrist) can be obtained, when necessary. However, the large majority of women with amenorrhea have relatively simple problems—polycystic ovarian syndrome (PCOS), hypothalamic amenorrhea, hyperprolactinemia, and ovarian failure—all of which can be managed easily by primary care clinicians.
The diagnostic evaluation described here is not new. With minor modifications, it has been applied successfully for several decades. Before describing the evaluation in detail, amenorrhea first must be defined, so as to identify the patients who warrant evaluation. A brief preliminary review of the physiologic mechanisms involved in menstruation provides the framework necessary to understand and follow the logical design of the diagnostic evaluation.
Definition of Amenorrhea
The age at which menarche should be expected varies with individual differences in the age at the onset of puberty. The normal pubertal progression is discussed in detail in Chaper 10 and is only briefly summarized here. In general, the first menses should occur within 2-3 years after the initiation of pubertal development. In most young girls (approximately 80%), the first sign of puberty is an acceleration of growth, followed by breast budding (thelarche), and the appearance of pubic hair (adrenarche). In the remainder (approximately 20%) adrenarche precedes thelarche by a brief interval, but the two events typically are closely linked. Consequently, menarche can occur as early as age 10 (when puberty begins at age 8), and rarely occurs later than age 16 (when puberty begins at age 13). On average, the mean ages for thelarche, adrenarche, and menarche in African-American girls are 6-12 months earlier than in Caucasian American girls. Once normal menstrual cycles have been established, they should occur at regular intervals ranging between 25 and 35 days. Therefore, patients fulfilling any of the following criteria should be evaluated for amenorrhea:
No menses by age 14 in the absence of growth or development of secondary sexual characteristics.
No menses by age 16 regardless of the presence of normal growth and development of secondary sexual characteristics.
In women who have menstruated previously, no menses for an interval of time equivalent to a total of at least three previous cycles, or 6 months.
Having affirmed the traditional definition of amenorrhea, it is important to point out that strict adherence to these criteria can result in improper management of individual patients. For example, there is no reason to defer the evaluation of a young girl who presents with the classical phenotype of Turner syndrome. Similarly, a 14-year-old girl who has no vagina should not be advised to return in 2 years. All patients deserve a considerate evaluation whenever their anxieties, or those of their parents, are brought to the attention of a clinician. Finally, the possibility of pregnancy always should be considered.
Traditionally, amenorrhea has been categorized as primary or secondary. Primary amenorrhea describes patients who never have menstruated and secondary amenorrhea describes those who have menstruated previously but now do not. The differential diagnoses of primary and secondary amenorrhea differ, but only to a limited extent. For example, the diagnosis of mullerian agenesis is possible only in patients with primary amenorrhea and premature menopause necessarily occurs only in women with secondary amenorrhea. However, besides helping to narrow the scope of diagnostic possibilities, the classical distinction between primary and secondary amenorrhea serves little practical purpose. Such preliminary categorization sometimes even can mislead the evaluation or its interpretation. In any case, the diagnostic approach recommended here can be applied effectively in all women with amenorrhea.
Basic Principles in Menstrual Function
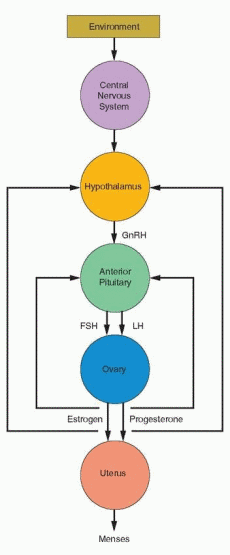
The clinical demonstration of menstrual function requires visible external evidence of the menstrual discharge. For that to occur, the genital outflow tract must be anatomically intact with continuous connection between the vaginal orifice, the vaginal canal, the endocervix, and the uterine cavity. The uterus also must contain a functional endometrium that can respond to the actions of ovarian sex steroid hormones, estrogen and progesterone,
across the ovarian cycle of follicle development, ovulation, and corpus luteum function. The ovaries must contain viable follicles that can respond to stimulation by the gonadotropins, follicle-stimulating hormone (FSH) and luteinizing hormone (LH), released from the anterior pituitary. In turn, pituitary gonadotropin secretion depends on the action of gonadotropin-releasing hormone (GnRH), secreted from the medial basal hypothalamus into the portal vascular network that bathes the anterior pituitary. Finally, the pulsatile pattern of hypothalamic GnRH secretion is governed by input from higher centers that interpret and translate environmental stimuli, and modulated by the feedback effects of ovarian sex steroids. The entire system is highly regulated by a complex mechanism that integrates biophysical and biochemical information composed of interacting hormonal signals, autocrine/paracrine factors, and target cell reactions.
across the ovarian cycle of follicle development, ovulation, and corpus luteum function. The ovaries must contain viable follicles that can respond to stimulation by the gonadotropins, follicle-stimulating hormone (FSH) and luteinizing hormone (LH), released from the anterior pituitary. In turn, pituitary gonadotropin secretion depends on the action of gonadotropin-releasing hormone (GnRH), secreted from the medial basal hypothalamus into the portal vascular network that bathes the anterior pituitary. Finally, the pulsatile pattern of hypothalamic GnRH secretion is governed by input from higher centers that interpret and translate environmental stimuli, and modulated by the feedback effects of ovarian sex steroids. The entire system is highly regulated by a complex mechanism that integrates biophysical and biochemical information composed of interacting hormonal signals, autocrine/paracrine factors, and target cell reactions.
The basic requirements for normal menstrual function thus include four anatomically and functionally distinct structural components—the genital outflow tract including the
uterus, the ovary, the pituitary, and the hypothalamus—thus providing a natural and useful hierarchy for organizing the diagnostic evaluation of amenorrhea. Accordingly, the many causes of amenorrhea can be categorized according to the site or level of the disorder or disturbance:
uterus, the ovary, the pituitary, and the hypothalamus—thus providing a natural and useful hierarchy for organizing the diagnostic evaluation of amenorrhea. Accordingly, the many causes of amenorrhea can be categorized according to the site or level of the disorder or disturbance:
Disorders of the genital outflow tract and uterus
Disorders of the ovary
Disorders of the anterior pituitary
Disorders of the hypothalamus or central nervous system
 |
Amenorrhea can result from congenital or acquired disease or dysfunction at any level in the system and can involve more than one mechanism. For example, PCOS involves a number of interrelated pathophysiologic mechanisms operating at the ovarian, pituitary and hypothalamic levels.
Evaluation of Amenorrhea
The evaluation of amenorrhea, like any other complaint, begins with a careful medical history and physical examination, which always provide valuable diagnostic clues. Information gained from the medical history and physical examination clearly can exclude certain diagnostic possibilities, but first impressions also can be deceiving and lead to errors in judgment and to inappropriate, costly, and needless testing. A methodical, systematic approach to diagnosis therefore is best.
Logically, the recommended evaluation for amenorrhea is designed to separate the reproductive system into its distinct structural components—the genital outflow tract and uterus, the ovary, the pituitary, and the hypothalamus—and to test the functional integrity of each, beginning at the lowest level and progressing systematically to the higher levels of the system until the cause is determined.
Medical History
The menstrual history is, of course, key. Primary amenorrhea speaks for itself, but cyclic pelvic or lower abdominal pain or urinary complaints can be caused by developmental anomalies resulting in obstructed menstrual flow (cryptomenorrhea), as may be caused by an imperforate hymen, transverse vaginal septum, or cervical atresia. In women with secondary amenorrhea, the history surrounding the onset of amenorrhea can provide important diagnostic clues. Onset following curettage or other uterine surgery clearly suggests the possibility of damage to the reproductive tract. The menstrual history in women having the most common causes of amenorrhea is distinctly different and easily recognized. Women with PCOS classically present with infrequent and irregular menses dating from menarche or early adulthood, and gradually progressive hirsutism. In most with hypothalamic amenorrhea, the onset of amenorrhea temporally relates to events resulting in severe nutritional, physical, or emotional stress. Women with hyperprolactinemia or premature ovarian failure commonly notice a gradual decrease in their regular intermenstrual interval, followed by increasing oligomenorrhea, and finally, amenorrhea, one sometimes accompanied by galactorrhea, and the other by hot flushes.
Questions relating to past medical history, general health, and lifestyle can identify a severe or chronic illness such as diabetes, renal failure, or inflammatory bowel disease, previous head trauma, or evidence of physical or psychological stress. Specific history relating to weight loss or weight gain and to the frequency and intensity of exercise is highly relevant and often revealing. Headaches, seizures, vomiting, behavioral changes, or visual symptoms may suggest a CNS disorder. Vaginal dryness or hot flushes are evidence of estrogen deficiency and suggest ovarian failure. Progressive hirsutism or virilization is an indication of hyperandrogenism that may relate to PCOS, non-classical (late-onset) congenital adrenal hyperplasia (CAH), or an androgen-producing tumor of the ovary or adrenal. Symptoms of galactorrhea obviously suggest hyperprolactinemia. History relating to the time and duration of any treatment with oral contraceptive pills (OCP), progestins (e.g., depot-medroxyprogesterone acetate), GnRH agonists, or other medications or drugs that can affect central neurotransmitter secretion (phenothizines, reserpine derivatives, amphetamines, benzodiazepines, antidepressants, dopamine antagonists, opiates) also can provide important diagnostic clues.
Physical Examination
The overall body habitus often provides important information. Height, weight, and body mass index (BMI) should be determined and recorded. Short stature (less than 60 inches) and sexual infantilism are hallmarks of gonadal dysgenesis. Low body weight frequently is associated with hypothalamic amenorrhea resulting from poor nutrition (eating disorders, malabsorbsion syndromes) or physical, psychological, or emotional stress. Obesity or an increased waist-hip ratio (>0.85) are common features of women with PCOS.
Examination of the skin can reveal a soft, moist texture as seen in hyperthyroidism; a rapid pulse, exopthalmos or lid lag, a fine tremor, and hyperreflexia suggest the diagnosis of Graves’ disease. Conversely, dry, thick skin, a slow pulse, diminished reflexes, and thinning of the hair suggest hypothyroidism. A goiter or thyroid nodule is further evidence of a thyroid disorder; both hypothyroidism and hyperthyroidism can be associated with amenorrhea. Orange discoloration of the skin, without scleral icterus, can result from hypercarotinemia associated with excessive ingestion of low calorie carotene-containing fruits and vegetables in dieting women. Acanthosis nigricans (velvety hyperpigmented skin observed most commonly at the nape of the neck, in the axillae, groin, and beneath the breasts) strongly suggests severe insulin resistance and the possibility of diabetes. Acne and hirsutism are indications of hyperandrogenism that may be associated with PCOS, non-classical CAH, or exposure to androgenic anabolic steroids. When accompanied by any sign of frank virilization (deepening of the voice, fronto-temporal balding, decrease in breast size, increased muscle mass, clitoromegaly), the possibility of ovarian hyperthecosis or an ovarian or adrenal neoplasm must be considered.
Examination of the breasts deserves careful attention. Breast development is a reliable indicator of estrogen production or exposure to exogenous estrogens. The Tanner stage of breast development should be noted (Chapter 10). A secondary arrest of breast development suggests a disruption of the hypothalamic-pituitary-ovarian (HPO) axis. When menarche has not followed breast development at the expected time, a developmental anomaly of the reproductive tract also should be considered. Breast examination should include gentle compression, beginning at the base and moving toward the nipple. Secretions that result from hormonal stimulation typically emerge from multiple duct openings in the nipple, whereas a discharge relating to breast pathology usually arises from a single duct. Microscopic examination of any expressed cloudy or white nipple secretions demonstrating lipid droplets confirms galactorrhea and suggests hyperprolactinemia.
Abdominal examination rarely may reveal a mass as may result from hematometra or an ovarian neoplasm. Growth of sexual hair in the infraumbilical region suggests hyperandrogenism. Abdominal striae raise the possibility of Cushing syndrome, but most often result from progressive obesity or previous pregnancy.
Careful examination of the external genitalia and lower genital tract is key. The presence of pubic hair growth reliably reflects androgen production or exposure. Because breast development and growth of pubic hair typically progress in a symmetrical manner, their Tanner stages should be consistent. Absent or scant growth of sexual hair can be expected in otherwise sexually infantile girls, but also is a classical sign of androgen insensitivity syndrome (AIS) when breast development is asymmetrically advanced. Attempts at office examination of the vagina in sexually infantile girls or those with a small hymeneal ring are generally unrewarding and often even counterproductive, but whenever feasible, speculum examination should be performed. A patent vagina and normal cervix excludes mullerian/vaginal agenesis, AIS, and obstructive causes of amenorrhea such as an imperforate hymen or transverse vaginal septum. In those with primary amenorrhea having an absent or infantile vaginal orifice, rectal examination should be performed to detect any distended hematocolpos that may form above the obstruction when the uterus is present and functional.
Evaluation of the Genital Outflow Tract and Uterus
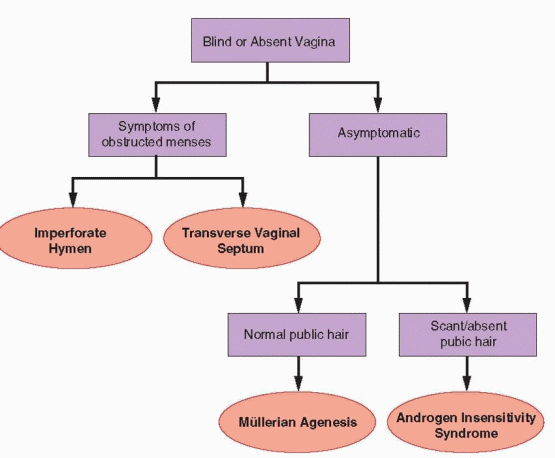
Evaluation of the genital outflow tract and uterus can be organized easily based on the menstrual history and physical examination of the genital anatomy. Primary amenorrhea with a blind or absent vagina points directly to a developmental anomaly of the genital outflow tract. Primary or secondary amenorrhea with a patent vagina and visible cervix excludes abnormalities of the genital outflow tract, except in those with history of previous cervical or uterine surgery or infection in whom the possibilities of cervical stenosis and intrauterine adhesions or other endometrial damage must be considered.
In premenarchial age girls with the incidental finding of an absent vaginal orifice, diagnosis can be more difficult, but also is seldom urgent. Although pelvic ultrasonography generally can determine whether a uterus is present, imaging must be interpreted cautiously because even abdominal/pelvic magnetic resonance imaging (MRI) can be misleading when the reproductive organs are immature and very small. Remaining alert to the diagnostic possibilities, careful observation over time is preferable to invasive investigations otherwise unnecessary in the asymptomatic prepubertal girl.
Abnormal Genital Anatomy
The embryology of the female genital tract is complex but generally well defined and is described in detail in Chapter 4. Briefly summarized, it involves both the medial migration and midline fusion of the müllerian (paramesonephric) ducts to form the uterus, cervix, and upper vagina, and the vertical fusion of that developing ductal system with the invaginating urogenital sinus that forms the lower vagina and the introitus. Outflow tract abnormalities that result from failure of müllerian duct development include vaginal/müllerian agenesis and AIS; the presence or absence of pubic hair distinguishes the two. Abnormalities caused by failure of vertical fusion include imperforate hymen and transverse vaginal septum or cervical atresia. Although all are uncommon and the clinician may have only limited or no previous experience with any of the four, each has unique and distinguishing features that, in most cases, point to the correct diagnosis at time of the initial visit. Only limited additional evaluation, described in a later section of this chapter, is required to firmly establish the diagnosis and to plan treatment.
Normal Genital Anatomy
In those with primary or secondary amenorrhea having a patent vagina and visible cervix, the likelihood of a genital outflow tract abnormality is very small. The only possibilities that need be considered are cervical stenosis and intrauterine adhesions (Asherman syndrome) or other endometrial damage that may result from surgical trauma or infection. Since these are acquired conditions, they result in secondary amenorrhea with an onset that typically correlates closely with the time of the previous insult. Both receive separate and thorough discussion in the later section of this chapter devoted to specific disorders of the genital outflow tract and uterus.
 |
Evaluation of Ovarian Function
In women with normal genital tract anatomy and no relevant history to suggest the possibility of cervical stenosis or Asherman syndrome, disorders of the genital outflow tract and uterus can be excluded and further stepwise evaluation is required to determine the cause of amenorrhea. Attention now may be focused on the next level of the reproductive system, the ovary.
Abnormalities of ovarian function are the most common overall cause of amenorrhea and include a wide variety of disorders ranging from simple chronic anovulation, as in women with PCOS, obesity, thyroid disorders and hyperprolactinemia, to complete ovarian failure relating to chromosomal abnormalities or other genetic disorders such as Fragile X (FMR1) premutations and galactosemia, autoimmune disease, radiation or chemotherapy. These and other specific causes of ovarian failure and the mechanisms involved are discussed at length in a later section of this chapter. The focus here is on the evaluation of ovarian function and on the diagnosis and treatment of common chronic anovulatory disorders.
The most obvious measure of ovarian function is estrogen production. Unfortunately, one cannot rely on symptoms and signs of estrogen deficiency to identify hypogonadal women.
Genitourinary atrophy develops only gradually and is not observed commonly in young women, even when estrogen levels are clearly low, and vasomotor symptoms typically are absent in women with hypothalamic dysfunction. Other methods for assessing the level of ovarian estrogen production include measurement of the serum estradiol concentration and “bioassays” based on clinical observations of the amount and character of cervical mucus, the results of a “progestin challenge test,” or measurement of endometrial thickness by transvaginal ultrasonography. Although each method is useful, each also has pitfalls, no one method is definitive, and more than one measure therefore is recommended. Overall, the duration of amenorrhea and other clinical history and features are as or more important and useful for assessing ovarian function.
Genitourinary atrophy develops only gradually and is not observed commonly in young women, even when estrogen levels are clearly low, and vasomotor symptoms typically are absent in women with hypothalamic dysfunction. Other methods for assessing the level of ovarian estrogen production include measurement of the serum estradiol concentration and “bioassays” based on clinical observations of the amount and character of cervical mucus, the results of a “progestin challenge test,” or measurement of endometrial thickness by transvaginal ultrasonography. Although each method is useful, each also has pitfalls, no one method is definitive, and more than one measure therefore is recommended. Overall, the duration of amenorrhea and other clinical history and features are as or more important and useful for assessing ovarian function.
Seum Estradiol Concentration
A serum estradiol measurement is easy to obtain, relatively inexpensive, and objective. Reasonably, one would expect to find normal estrogen levels in women with normal ovaries whose amenorrhea results simply from mild dysregulation and chronic anovulation, as in obese women and those with PCOS, and to find low estrogen levels in women with ovarian failure, pituitary disease, or more severe hypothalamic dysfunction. Unfortunately, serum estradiol concentrations can fluctuate erratically in all conditions, normal or low on any given day, and therefore can be misleading. A random estradiol concentration greater than approximately 40 pg/mL clearly suggests the presence of functional ovarian follicles but also is common during a premature or normal perimenopause and occurs sporadically in women with hypothalamic amenorrhea. A low random estradiol concentration may suggest ovarian failure, but also is typical of women with hypothalamic amenorrhea and may be observed in those with less severe forms of chronic anovulation, as in normal women during the early follicular phase.
Bioassays of Estrogen Production
The observation of “estrogenic” cervical mucus—clear, watery, and relatively abundant— suggests a normal level of ovarian estrogen production, but its absence cannot be interpreted confidently because many normal women exhibit such mucus only during the late follicular phase of the cycle when estrogen levels are relatively high, or not at all.
The progestin challenge test is based on the premise that progestin treatment (e.g., medroxyprogesterone acetate 10 mg daily for 5-7 days, or progesterone in oil 200 mg i.m.) will induce menses only in those having normal circulating estrogen concentrations. A pure progestational agent must be used because endogenous estrogen status cannot be inferred from the response to an OCP that contains both estrogen and progestin. The more potent synthetic progestins such as medroxyprogesterone acetate are a better choice than oral micronized progesterone, which must be administered in relatively high doses (e.g., 300 mg daily) to achieve a response.1 A positive test—bleeding within 2-7 days after completion of progestin treatment—implies normal estrogen production and ovarian function, and a negative test—no withdrawal menses—suggests hypogonadism. Scant withdrawal bleeding or spotting suggests marginal levels of endogenous estrogen production. However, the overall correlation between withdrawal bleeding and estrogen status is far from perfect; both false positive (withdrawal bleeding despite generally low levels of estrogen production) and false negative results (absent bleeding despite significant estrogen production) are relatively common. Up to 40-50% of women whose amenorrhea relates to stress, exercise, weight loss, hyperprolactinemia, or ovarian failure, in whom estrogen levels generally are low, exhibit withdrawal bleeding.2,3 Up to 20% of amenorrheic women
with significant estrogen production have no withdrawal bleeding,4 in some because the endometrium is decidualized by high circulating androgen levels.
with significant estrogen production have no withdrawal bleeding,4 in some because the endometrium is decidualized by high circulating androgen levels.
The endometrial thickness, determined by transvaginal ultrasonography (the maximum 2-layer thickness in the mid-sagittal plane), is a measure of endometrial proliferation, which reflects the level of estrogen production. Endometrial thickness correlates with both the serum estradiol concentration and with the response to a progestin challenge in women with amenorrhea. In one study involving 44 women with secondary amenorrhea, endometrial thickness was significantly greater in 32 women who had withdrawal bleeding (10.3±4.1 mm) than in 12 who did not (5.0±1.3 mm); the serum estradiol level also was significantly greater (45.3±19.4 vs. 18.6±8.0 pg/mL), and an endometrial thickness measuring 6.0 mm or greater predicted withdrawal bleeding with 95% accuracy.2 An added potential benefit of endometrial thickness as a measure of ovarian estrogen production is that it can help to identify individuals with chronic anovulation at low risk for having associated pathology such as hyperplasia or cancer.
Serum FSH Concentration
The serum FSH concentration is another obvious and useful, but indirect, measure of ovarian function. A normal or low serum FSH level indicates the presence of functional ovarian follicles and may be observed in a variety of conditions associated with amenorrhea, including chronic anovulation (e.g., PCOS), pituitary disease, and hypothalamic dysfunction. A high serum FSH concentration is a reliable indication of ovarian follicular depletion or failure. Exceptions are rare and include inactivating mutations involving the FSH or LH receptor, enzyme deficiencies (17a-hydroxylase, aromatase), and functional pituitary and ectopic FSH-secreting tumors. Because the clinical implications of an elevated FSH level are serious, one or more repeated measurements are warranted to confirm the finding.
Clinical State | Serum FSH | Serum LH |
Normal adult female | 5-20 IU/L (midcycle peak ∽ 2 times | 5-20 IU/L (midcycle peak ∽ 3 times |
Hypogonadotropic state: Prepubertal, | <5 IU/L | <5 IU/L |
Hypergonadotropic state: Postmenopausal, | >20 IU/L | >40 IU/L |
Although certainly not inappropriate, generally it is not necessary or helpful to also measure the serum LH concentration because levels of the two gonadotropins typically move in parallel. The one notable and highly relevant exception in women with amenorrhea—the “monotropic” rise in FSH that signals a more advanced stage of follicular depletion—can be detected by measuring FSH alone. A moderately increased serum LH concentration frequently is observed in women with PCOS, but is not a diagnostic criterion and has no other clinical relevance. A “reversed” LH/FSH ratio (LH lower than FSH), like that seen in prepubertal girls, suggests but does not prove hypothalamic dysfunction. During the midcycle gonadotropin surge in ovulatory cycles, LH levels increase more than those of FSH, but that has little relevance in women with amenorrhea. Other conditions in which levels of the two gonadotropins diverge significantly are truly rare and include ectopic gonadotropin secretion by tumors outside the reproductive tract, single gonadotropin deficiencies resulting from mutations in genes encoding the b-subunit of LH or FSH, and the very rare functional gonadotroph adenoma that secretes clinically important amounts of one gonadotropin (FSH) but not the other.
Measurement of the serum FSH level traditionally has been recommended only for those having demonstrable evidence of hypogonadism (e.g., a negative progestin challenge), as the means to differentiate patients with gonadal failure from those with hypothalamic or pituitary causes of amenorrhea. However, routine measurement of serum FSH in the evaluation of amenorrhea is not difficult to justify because none of the available measures of ovarian estrogen production is completely reliable, for reasons already described. A serum FSH contributes to a more confident clinical assessment of ovarian function and helps differentiate patients with common chronic anovulatory conditions from those with more severe hypogonadism who otherwise may go unrecognized and who require further specific evaluation, counseling, or treatment. For example, when evaluation suggests marginal levels of estrogen production (e.g., serum estradiol 30-40 pgmL or scant bleeding after a progestin challenge), a low serum FSH can identify those who merit further evaluation to exclude pituitary and hypothalamic disease, as described below. Conversely, in women with normal levels of estrogen production, a moderately elevated FSH level (e.g., 10-15 IU/L) can reveal a diminished ovarian reserve in women who may be afforded the chance to actively pursue their reproductive goals before the opportunity is lost, if alerted to their advanced reproductive aging. Like the other measures of ovarian function, the serum FSH concentration must be interpreted carefully, in its clinical context. FSH levels can fluctuate unpredictably, particularly during the years immediately preceding the menopause, regardless whether it occurs prematurely or at the usual age.
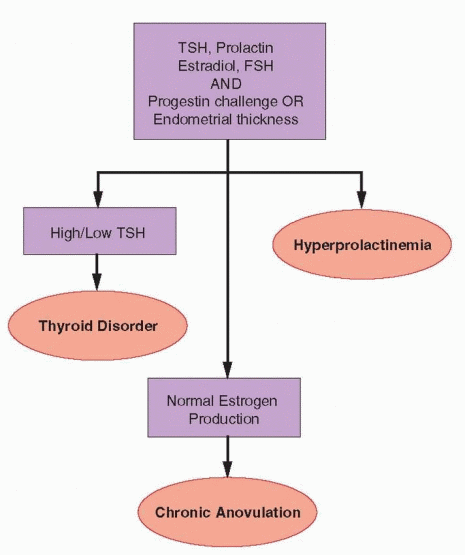
Chronic Anovulation
When evaluation reveals clear evidence of normal ovarian estrogen production and the serum FSH level also is normal, the diagnosis of chronic anovulation is established. Hyperprolactinemia is one of the most common causes of anovulation and amenorrhea and, although less common, thyroid disorders are easily identified and treated. Measurement of the serum prolactin and thyroid-stimulating hormone (TSH) concentrations are therefore justified in all women with amenorrhea. For efficiency, both can be measured along with the serum FSH and estradiol levels, at the outset of the evaluation. When all are normal, no further evaluation is required.
Besides thyroid and prolactin disorders, common and likely causes of chronic anovulation include PCOS, obesity, stress or exercise, and reproductive aging. In all but the last, anovulation can be attributed to a dysfunctional HPO axis in which gonadotropin secretion is sufficient to stimulate follicular development and estrogen production, but the system lacks the coordination required to achieve ovulation. Women with classical PCOS usually are easily recognized because they also exhibit signs of hyperandrogenism, unlike most women whose chronic anovulation relates solely to weight gain or obesity; the pathophysiology of the two disorders is complex and is discussed at length in Chapter 12 (PCOS) and Chapter 19 (obesity). Severe hirsutism or signs of virilization warrant additional specific evaluation to exclude enzyme deficiencies, androgen-secreting tumors, and Cushing’s syndrome, as described in Chapter 13. The diagnosis of anovulation relating to emotional, nutritional, or physical stress is made by exclusion, but often is suggested by the medical history and physical examination. The management of chronic anovulation associated with thyroid disorders and hyperprolactinemia is summarized here.
The newest ultra-sensitive TSH assays now in common use detect both primary hypothyroidism (elevated TSH) and primary hyperthyroidism (low TSH); either may result in chronic anovulation and amenorrhea. Although only a few patients presenting with amenorrhea will have a thyroid disorder that is not clinically apparent, their exclusion and
treatment are so simple that routine measurement of TSH is justified; a return of ovulatory cycles typically follows the restoration of normal thyroid hormone levels. Any abnormal TSH value should be confirmed and accompanied by measurement of serum thyroxine (tetra-iodothyronine; T4, or free T4) to better define the nature and extent of the thyroid disorder. An elevated TSH with a normal free T4 concentration indicates a subclinical hypothyroidism, best viewed as a compensated state wherein normal levels of T4 are maintained, but only under increased levels of pituitary stimulation. Although observation and periodic re-evaluation are reasonable in patients with subclinical hypothyroidism, because not all will develop frank hypothyroidism, treatment is warranted in those with menstrual dysfunction or infertility. In those with a low TSH and a normal free T4 level, serum tri-iodothyronine (T3) should be measured; an elevated T3 can identify hyperthyroidism that otherwise might escape detection. When the T3 also is normal, a subclinical hyperthyroidism is likely and should be followed carefully. On rare occasions, both TSH and free T4 levels are low, suggesting a secondary hypothyroidism of pituitary origin that requires additional evaluation to determine the cause and whether other pituitary functions also are affected.
treatment are so simple that routine measurement of TSH is justified; a return of ovulatory cycles typically follows the restoration of normal thyroid hormone levels. Any abnormal TSH value should be confirmed and accompanied by measurement of serum thyroxine (tetra-iodothyronine; T4, or free T4) to better define the nature and extent of the thyroid disorder. An elevated TSH with a normal free T4 concentration indicates a subclinical hypothyroidism, best viewed as a compensated state wherein normal levels of T4 are maintained, but only under increased levels of pituitary stimulation. Although observation and periodic re-evaluation are reasonable in patients with subclinical hypothyroidism, because not all will develop frank hypothyroidism, treatment is warranted in those with menstrual dysfunction or infertility. In those with a low TSH and a normal free T4 level, serum tri-iodothyronine (T3) should be measured; an elevated T3 can identify hyperthyroidism that otherwise might escape detection. When the T3 also is normal, a subclinical hyperthyroidism is likely and should be followed carefully. On rare occasions, both TSH and free T4 levels are low, suggesting a secondary hypothyroidism of pituitary origin that requires additional evaluation to determine the cause and whether other pituitary functions also are affected.
A few women with hypothyroidism will develop a secondary hyperprolactinemia and even galactorrhea. The likelihood of hyperprolactinemia increases with the duration of hypothyroidism; galactorrhea is more common in young women with higher prolactin levels.5 The mechanism probably involves both the gradual depletion of hypothalamic dopamine (the putative prolactin-inhibiting factor) and constant stimulation of pituitary lactotropes by thyrotropin-releasing hormone (TRH), which may cause pituitary hypertrophy or hyperplasia and sometimes even enlargement or erosion of the sella turcica.6,7 Although hormone levels rapidly normalize with appropriate treatment, the disappearance of breast secretions in those with galactorrhea is gradual and can take several months. Patients with primary hypothyroidism and hyperprolactinemia may present with either primary or secondary amenorrhea.8
 |
Hyperprolactinemia
Hyperprolactinemia is among the most common causes of secondary amenorrhea and also may result in delayed puberty and primary amenorrhea when it arises before menarche. A serum prolactin concentration is therefore justified in all women with amenorrhea. A normal random measurement (<15-20 ng/mL in most clinical laboratories) excludes hyperprolactinemia. The prolactin level is fairly stable throughout the day but can increase transiently during sleep, exercise, breast stimulation, and meals. To avoid otherwise unnecessary and costly imaging, mildly elevated prolactin levels (20-40 ng/mL) are best repeated and confirmed before the diagnosis of hyperprolactinemia is made.
The mechanism by which hyperprolactinemia results in anovulation and amenorrhea relates to a disruption or inhibition of the normal hypothalamic GnRH pulse rhythm, resulting in ineffective or frankly low levels of gonadotropin secretion. It may be that increased circulating prolactin levels stimulate a generalized increase in hypothalamic dopaminergic neuronal activity, intended to suppress prolactin secretion but also inhibiting GnRH neurons. In any case, the end result is anovulation or an even more profound hypogonadotropic hypogonadism, depending on the extent to which gonadotropin secretion is suppressed. Mild hyperprolactinemia (20-50 ng/mL) may cause only a short luteal phase, resulting from poor preovulatory follicular development.9,10 Moderate hyperprolactinemia (50-100 ng/mL) frequently causes oligomenorrhea or amenorrhea, and higher prolactin levels (>100 ng/mL) typically result in frank hypogonadism with low estrogen levels, and their clinical consequences (e.g., genitorurinary atrophy, osteopenia).11,12
The symptom or finding of galactorrhea cannot reliably identify those whose amenorrhea results from hyperprolactinemia. Only about one-third of women with hyperprolactinemia exhibit galactorrhea, probably because breast milk production requires estrogen and hyperprolactinemia often results in anovulation or a more severe secondary hypogonadotropic hypogonadism and low circulating estrogen levels. The structural heterogeneity of prolactin offers another possible explanation. Prolactin circulates in various forms that have varying bioactivity (manifested by galactorrhea) and immunoactivity (recognition by immunoassay).13,14 and 15 The predominant form (80-95%) is monomeric (molecular weight 23 kDa), which is more biologically active than larger glycosylated forms that may combine to form dimmers or trimers (“big prolactin,” 50-60 kDa) and other even larger varieties (macroprolactin, >100 kDa), which result from the aggregation of smaller prolactin molecules bound together with immunoglobulins.16 The larger molecular forms are cleared more slowly, predominate in women with hyperprolactinemia having normal menses, and result in minimal or no galactorrhea.17 If suspected, the diagnosis of macroprolactinemia can be confirmed by asking the laboratory to pretreat the patient’s serum with polyethylene glycol to precipitate the macroprolactin before performing the prolactin assay.18 In women with mildly elevated prolactin levels, diagnosis of macroprolactinemia avoids unnecessary and costly imaging aimed at excluding pituitary and hypothalamic mass lesions.
Hyperprolactinemia has many causes that are discussed in detail in the chapter dedicated to the breast (Chapter 16) and briefly summarized here.19 Hyperprolactinemia may result from hypothyroidism, prolactin-secreting pituitary adenomas, and other pituitary or hypothalamic tumors that may compress the pituitary stalk and disrupt the delivery of dopamine. A variety of drugs that lower dopamine levels or inhibit dopamine action may cause hyperprolactinemia, including amphetamines, benzodiazepines, butyrophenones, metaclopromide, methyldopa, opiates, phenothiazines, reserpine, and tricyclic antidepressants. Breast or chest wall surgery, cervical spine lesions, or herpes zoster (affecting the dermatome that includes the breast) may activate the afferent sensory neural pathway that stimulates prolactin secretion in a manner similar to suckling. Renal insufficiency and macroprolactinemia may cause hyperprolactinemia, due to decreased clearance. Rarely, hyperprolactinemia may result from ectopic prolactin secretion by pituitary tissue in the pharynx, by bronchogenic and renal-cell carcinomas, or by a gonadoblastoma or prolactinoma that may
arise in benign or malignant ovarian teratomas.20,21,22,23 and 24 All possible causes must be considered and excluded; a careful history can eliminate most of the possibilities. When the cause reasonably may be attributed to a medication, a trial discontinuation or use of an alternative drug should be considered, in consultation with the prescribing physician. When that is not possible, further evaluation to exclude a pituitary or hypothalamic mass lesion is required.
arise in benign or malignant ovarian teratomas.20,21,22,23 and 24 All possible causes must be considered and excluded; a careful history can eliminate most of the possibilities. When the cause reasonably may be attributed to a medication, a trial discontinuation or use of an alternative drug should be considered, in consultation with the prescribing physician. When that is not possible, further evaluation to exclude a pituitary or hypothalamic mass lesion is required.
Women with amenorrhea and hyperprolactinemia that cannot be attributed confidently to medication or another specific cause require further evaluation with imaging to exclude pituitary tumors and hypothalamic mass lesions. (see Evaluation of Pituitary Function, below). Pituitary adenomas and their management are discussed in detail in a later section of this chapter devoted specifically to pituitary causes of amenorrhea. Discussion here is limited to the treatment of hyperprolactinemia unassociated with any demonstrable sellar abnormality.
Treatment with a dopamine agonist restores ovulatory function and menses within several weeks in the large majority of women with hyperprolactinemia. Although the amount of breast secretions in those with galactorrhea decreases significantly over the same interval of time, complete cessation often takes considerably longer.25 Both bromocriptine and cabergoline are highly effective. Bromocriptine has a relatively short half life, must be administered daily (at bedtime) or twice daily, and often is associated with gastrointestinal side effects such as nausea. Cabergoline is a selective dopamine receptor type 2 agonist having fewer side effects than bromocriptine, greater potency and a longer duration of action requiring less frequent administration (twice weekly), and can be effective in those who cannot tolerate or prove resistant to bromocriptine.26,27 However, cabergoline also has been associated with hypertrophic valvular heart disease when used in high doses (>3 mg daily) as in patients with Parkinson’s disease; mitogenic stimulation of normally quiescent valve cells via activation of serotonin receptors is the suspected mechanism.28,29 Although the doses required for effective treatment of hyperprolactinemia are much lower, longterm use of even relatively low doses may increase the risk of valvular heart disease.30,31 Consequently, cabergoline should be used in the lowest dose required to normalize serum prolactin concentrations and a trial discontinuation of treatment should be attempted if prolactin levels have been normal for 2 or more years.32 The dose of dopamine agonist treatment should be adjusted according to response, beginning with a low dose and increasing gradually as needed to normalize prolactin levels. In those who cannot tolerate oral treatment, vaginal administration is effective and associated with fewer size effects.33,34 Either drug may be used in women planning to conceive since both appear to be safe in early pregnancy.35,36
Unfortunately, amenorrhea and galactorrhea often promptly recur within weeks after discontinuation of dopamine agonist treatment and most therefore require long-term therapy. Treatment with a dopamine agonist is the obvious choice when the objective is ovulation induction and pregnancy or the elimination of troublesome galactorrhea. However, for those with neither specific indication, alternative treatments deserve careful consideration.
Although treatment with a dopamine agonist certainly is a logical choice, it is by no means the only choice or necessarily the best choice for all women with hyperprolactinemia and amenorrhea. It is important to remember that treatment should be focused on the patient, and not on the prolactin level. Hyperprolactinemia itself poses no particular health risks. In women not at risk for an unwanted pregnancy, cyclic progestin therapy will prevent the clinical consequences of chronic unopposed estrogen exposure in those who are not frankly hypogonadal, and in those who are, physiologic cyclic or combined estrogen/progestin treatment will prevent the consequences of chronic estrogen deficiency. In women who need contraception, treatment with a low-dose oral contraceptive achieves the same goals. In the past, treatment with exogenous estrogen was considered contraindicated for women with hyperprolactinemia due to fear it might aggravate the underlying pathophysiology or promote growth of a pituitary tumor, but experience has shown that
hormone therapy and oral contraceptives pose no such risks.37,38 The same treatments are useful in the management of women with medication-induced hyperprolactinemia and hypogonadism when the drug cannot be discontinued or another substituted. Dopamine agonists are best avoided in patients with medication-induced hyperprolactinemia, because they may interfere with or counteract the dopamine antagonist properties of their primary treatment.
hormone therapy and oral contraceptives pose no such risks.37,38 The same treatments are useful in the management of women with medication-induced hyperprolactinemia and hypogonadism when the drug cannot be discontinued or another substituted. Dopamine agonists are best avoided in patients with medication-induced hyperprolactinemia, because they may interfere with or counteract the dopamine antagonist properties of their primary treatment.
General Management
All patients with chronic anovulation require management, and with the limited evaluation described here, treatment can be implemented immediately. Clinicians are keenly aware that normal endometrium can progress to hyperplasia, atypia, and cancer within a relatively short interval of time. However, too often they believe the problem is relevant only in older aged women. The critical factor is not age, but the duration of exposure to unopposed estrogen stimulation. Young women who remain anovulatory for long periods of time can, and do, develop endometrial cancer.39,40,41 and 42 Although endometrial sampling is not indicated for all women with chronic anovulation, it should be considered seriously for those at greatest risk for endometrial pathology. Obese women and those with PCOS are the most likely candidates because obesity, hyperinsulinemia, and hyperandrogenism are known risk factors for endometrial neoplasia.43,44 Screening by endometrial thickness generally has poor positive predictive value for detecting endometrial pathology, but may be useful for identifying individuals at very low risk in whom biopsy safely may be omitted. No studies correlating endometrial thickness and histology in premenopausal women with amenorrhea have been performed. However, in premenopausal women with abnormal uterine bleeding, no serious pathology was found in those having an endometrial thickness less than 8 mm,45 and in asymptomatic postmenopausal women, an endometrial thickness less than 5-6 mm has greater than 99% negative predictive value for endometrial disease.46,47 Whereas some studies have suggested that amenorrheic women also may be at increased risk for developing breast cancer,48 the weight of available evidence suggests an inverse association between breast cancer risk and chronic anovulation (discussed in Chapter 16).49
At a minimum, women with chronic anovulation require periodic treatment with a progestin, to induce predictable menses and protect against the risk of developing endometrial pathology. For example, medroxyprogesteone acetate 5-10 mg daily can be administered for the first 12-14 days of each, or at least alternate, months; experience with varying hormone treatment regimens has demonstrated that treatment for an interval greater than 10 days is required to effectively counteract the growth-promoting effects of continuous estrogen exposure. It is important to note that cyclic treatment with a progestin, at physiologic doses, does not change the intrinsic rhythm of the HPO axis and will not prevent a sporadic ovulation. Therefore, if menses do not occur at the expected time, pregnancy must be considered and excluded. Absent bleeding after a course of progestin treatment also may indicate that estrogen production has fallen to grossly low levels and signal the need for further evaluation, as described in the section that follows. When reliable contraception is required, cyclic treatment with a low-dose oral contraceptive pill or a vaginal contraceptive ring is the obvious and better choice. There is no evidence that hormonal contraception has any impact, positive or negative, on menstrual cyclicity after treatment is discontinued.
For women with chronic anovulation having pregnancy as their goal, treatment should be aimed at inducing normal ovulatory cycles. Methods for ovulation induction are described in detail in Chapter 31. For women with thyroid disorders, specific treatment to restore normal thyroid function is indicated. For those with hyperprolactinemia, a dopamine agonist is the treatment of choice. Most having neither will respond to treatment with clomiphene citrate, reserving exogenous gonadotropin stimulation for those who do not.
Ovarian Failure
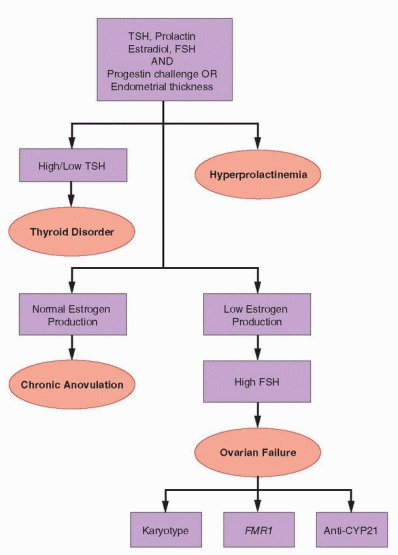
When evaluation reveals clear evidence of low ovarian estrogen production and the serum FSH level is consistently high, the diagnosis of ovarian failure is established.
Although premature follicular depletion is the cause in almost all cases, additional specific evaluation is indicated to exclude chromosomal and other genetic abnormalities and autoimmune disease that may have important potential health implications for the patient and other members of her family. The elements and purpose of the expanded diagnostic evaluation are summarized here. The known causes of ovarian failure and the disorders deserving specific consideration and exclusion are discussed at greater length in a later section of this chapter.
Karyotype
In all patients under age 30 with a diagnosis of ovarian failure, a karyotype should be obtained to exclude chromosomal translocations, deletions, and mosaicism that might offer an obvious explanation. A karyotype also identifies those having a Y chromosome in whom gonadectomy is indicated due to the significant risk for malignant transformation in occult testicular elements (20-30%). Signs of virilization cannot reliably identify the subset of women at risk because many having a Y chromosome exhibit no signs of excess androgen production. In women over age 30, ovarian failure reasonably can be regarded as premature menopause. Karyotype after age 30 generally is unnecessary because most tumors in patients with a Y chromosome arise before age 20, and virtually all before the age of 30.50,51 After age 30, women with short stature or a family history of early menopause still merit a karyotype to exclude X chromosome deletions and translocations that may affect other family members.52,53,54 and 55 Otherwise, pelvic ultrasonography can exclude the rare tumor not recognized previously.
Fragile X (FMR1) Premutations
Fragile X syndrome is the most common inherited cause of mental retardation and autism and results from abnormal expansion of an unstable trinucleotide (CGG) repeat sequence in the FMR1 (Fragile X Mental Retardation) gene, located on the long arm of the X chromosome (Xq27.3). The gene normally contains about 30 CGG repeats, but in those with Fragile X syndrome, the number exceeds 200. Convincing evidence has demonstrated an association between premature ovarian failure (POF) and fragile X “premutations,” characterized by 55-200 CGG repeats. Whereas the full mutation silences the FMR1 gene, resulting in little or no production of the corresponding mRNA or gene product (fragile X mental retardation protein, FMRP), the POF associated with premutations may reflect FMR1 mRNA gain-of-function toxicity.56 Women with premutations often exhibit endocrine evidence of early ovarian aging and up to one-third have an early menopause. The prevalence of premutations is approximately 14% in women with familial POF, and between 1% and 7% in sporadic cases of POF.56,57 Women with POF therefore should be offered testing for FMR1 premutations.58 Women who carry Fragile X premutations (and any also affected children or siblings) are at risk for having a child with Fragile X syndrome, because the length of the CGG repeat sequence is unstable and may expand to a full mutation when passed from mother to offspring. The inheritance and the implications of premutations are complex and affected women therefore should receive formal genetic counseling.
Autoimmune Screening
Ovarian failure sometimes may be the consequence of autoimmune disease.59 Addison’s disease (autoimmune adrenocortical insufficiency) has the strongest association with POF; the presence of autoantibodies to steroid-producing cells and observations of a lymphocytic infiltrate in the ovaries of affected individuals suggests the mechanism (autoimmune
oophoritis).60 The prevalence of other autoimmune diseases (e.g., thyroid autoimmunity, Type I diabetes, and myasthenia gravis) is higher among women with POF than in the general population, but there is no direct or compelling evidence to indicate a cause and effect relationship. Autoimmune ovarian failure generally occurs as part of a specific autoimmune polyendocrine syndrome (APS) that includes adrenal insufficiency. However, because POF may precede onset of adrenal insufficiency by several years, the autoimmune cause may not be recognized when the diagnosis of POF is first made.59 Women with POF should be tested for anti-adrenal antibodies (most easily demonstrated against the 21-hydroxylase enzyme, CYP21), and for anti-thyroid antibodies (anti-thyroid peroxidase and anti-thyroglobulain antibodies). The presence of anti-adrenal antibodies strongly implies autoimmune oophoritis as the cause of POF and idenifies women who should be carefully evaluated and followed to exclude adrenal insufficiency. The presence of thyroid autoantibodies does not prove autoimmune ovarian failure, but identifies women at risk for developing autoimmune thyroid disorders. Routine screening for other autoimmune endocrine disorders is unnecessary and can be reserved for those with clinical indications.61
oophoritis).60 The prevalence of other autoimmune diseases (e.g., thyroid autoimmunity, Type I diabetes, and myasthenia gravis) is higher among women with POF than in the general population, but there is no direct or compelling evidence to indicate a cause and effect relationship. Autoimmune ovarian failure generally occurs as part of a specific autoimmune polyendocrine syndrome (APS) that includes adrenal insufficiency. However, because POF may precede onset of adrenal insufficiency by several years, the autoimmune cause may not be recognized when the diagnosis of POF is first made.59 Women with POF should be tested for anti-adrenal antibodies (most easily demonstrated against the 21-hydroxylase enzyme, CYP21), and for anti-thyroid antibodies (anti-thyroid peroxidase and anti-thyroglobulain antibodies). The presence of anti-adrenal antibodies strongly implies autoimmune oophoritis as the cause of POF and idenifies women who should be carefully evaluated and followed to exclude adrenal insufficiency. The presence of thyroid autoantibodies does not prove autoimmune ovarian failure, but identifies women at risk for developing autoimmune thyroid disorders. Routine screening for other autoimmune endocrine disorders is unnecessary and can be reserved for those with clinical indications.61
 |
Evaluation of Pituitary Function
The normal feedback relationship between ovarian estrogen production and pituitary gonadotropin secretion dictates that low estrogen levels should cause a compensatory increase in FSH release to stimulate ovarian follicular development and estrogen secretion, just as they do during the early follicular phase of the normal cycle. When estrogen production is abnormally low, a low serum FSH concentration (<5 IU/L) indicates that inadequate or ineffective gonadotropin secretion is the cause and that even basic central feedback mechanisms in the HPO axis are not functioning. When estrogen levels are clearly low, a serum FSH level in the low normal range (5-10 IU/L) has the same interpretation and clinical implication, for two reasons. First, because the FSH level should be high when estrogen production is grossly low, even a “normal” value is, in fact, abnormally low in that clinical context. Second, although the measured level of immunoreactive FSH may be normal, the level of biologically active FSH clearly is not; if it were, follicular growth and estrogen production would be maintained. The biological activity of glycoprotein hormones varies with their carbohydrate moieties (discussed in Chapter 2) and evidence indicates that women with hypogonadotropic hypogonadism may secrete gonadotropins having altered patterns of glycosylation and reduced biological activity.62 Indeed, most women with hypogonadotropic hypogonadism have normal serum gonadotropin concentrations; extremely low or undetectable gonadotropin levels typically are observed only in those with large pituitary tumors or in patients with anorexia nervosa.
Imaging
When there is no clear explanation for hypogonadotropic hypogonadism (e.g., significant physical, nutritional, or emotional stress) or for hyperprolactinemia (e.g., medications), further evaluation with imaging is indicated to exclude tumors and to help distinguish between pituitary and hypothalamic causes. The method of choice is MRI (with gadolinium contrast) because it is more sensitive and accurate than other imaging techniques for detection of abnormalities within and near the sella turcica.63 MRI can demonstrate the nearby optic chiasm and also can detect blood, allowing hemorrhage and vascular abnormalities to be distinguished from other sellar mass lesions. Most sellar masses are pituitary adenomas, which account for 10% of all intracranial neoplasms. Other less common mass lesions in or near the sella include benign tumors (craniopharyngioma, hamartoma, meningioma), pituitary hyperplasia (thryotroph or gonadotroph hyperplasia due to long-standing primary hypothyroidism or gonadal failure), malignant tumors (germ cell, sarcoma, chordoma, carcinoma, lymphoma), metastases (lung, breast), cysts (Rathke’s cleft, arachnoid, dermoid), pituitary abscess, lymphocystic hypophpysitis, sarcoidosis, tuberculosis, and carotid arteriovenous fistula.
Although mass lesions are the most obvious abnormality to be excluded, other rare possibilities include Sheehan syndrome (pituitary infarct resulting from hypotension associated with postpartum hemorrhage), infiltrative hemosiderosis relating to frequent transfusions or hereditary hemochromatosis, traumatic brain injury,64 and mutations in the GnRH receptor.65
In the absence of any demonstrable mass lesion in the sellar region or relevant history suggesting another specific cause for pituitary damage, there is no need to perform any additional specific pituitary function tests. The clinical signs and symptoms associated with different types of functional and nonfunctional pituitary tumors and other specific pituitary causes of gonadotropin deficiency are discussed in a later section of this chapter.
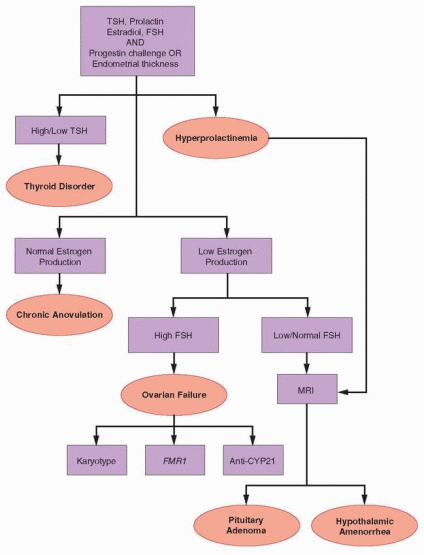 |
Evaluation of Hypothalamic Function
When imaging reveals no mass lesion and there is no reason to suspect other specific pituitary pathology, the diagnosis is functional hypothalamic amenorrhea, by exclusion. The pathophysiology of the disorder relates to a suppressed or otherwise abnormal pattern of pulsatile hypothalamic GnRH secretion, resulting in decreased gonadotropin secretion, absent follicular development, anovulation, and low serum concentrations of estradiol. The serum FSH (and LH, if measured) concentration is low or in the normal range; often, but not always, the FSH level is higher than LH as in prepubertal girls.
Unfortunately, there is no simple way to test, manipulate, or measure hypothalamic function to prove a GnRH deficiency. Whereas one might anticipate that the LH response to a
bolus of exogenous GnRH (e.g., 100 mg, administered subcutaneously) would be revealing, experience has shown it may be normal (>10 IU/L) or low in women with pituitary or hypothalamic disease. The response to repeated bolus GnRH administration (e.g., 24 hours after the first) may be somewhat more informative, due to the self-priming effect that GnRH has on its own receptor.66 The administration of exogenous pulsatile GnRH using a programmable infusion pump can restore normal gonadotropin secretion and menstrual function and induce ovulation in women with hypothalamic amenorrhea,67,68 but the considerable costs and logistical challenges of such treatment make it impractical and impossible to justify as a diagnostic test.
bolus of exogenous GnRH (e.g., 100 mg, administered subcutaneously) would be revealing, experience has shown it may be normal (>10 IU/L) or low in women with pituitary or hypothalamic disease. The response to repeated bolus GnRH administration (e.g., 24 hours after the first) may be somewhat more informative, due to the self-priming effect that GnRH has on its own receptor.66 The administration of exogenous pulsatile GnRH using a programmable infusion pump can restore normal gonadotropin secretion and menstrual function and induce ovulation in women with hypothalamic amenorrhea,67,68 but the considerable costs and logistical challenges of such treatment make it impractical and impossible to justify as a diagnostic test.
In most cases, the probable cause of hypothalamic amenorrhea can be identified, such as extreme emotional stress, acute weight loss or chronic malnutrition, or strenuous physical exercise. However, in others with hypothalamic amenorrhea, no obvious cause or precipitating event can be identified. Rare individuals with idiopathic hypogonadotropic hypogonadism may present with primary amenorrhea and infantile sexual development due to a congenital GnRH deficiency, resulting from the failure of GnRH neuronal development during embryogenesis or from mutations in the GnRH receptor, but specific evaluation to identify such abnormalities is not clinically necessary or indicated, except perhaps when other family members are affected. The causes of hypothalamic amenorrhea and its management are discussed at length in a later section of this chapter devoted to specific disorders of the hypothalamus.
Specific Causes of Amenorrhea
With only modest effort, time, and expense, the problem of amenorrhea has been dissected by systematic evaluation of the organ systems involved in menstrual function—the genital outflow tract and uterus, the ovary, the anterior pituitary, and the hypothalamus. Once the anatomic level of the disorder has been so defined, attention can turn to making a specific diagnosis. This section of the chapter considers each of the major causes of amenorrhea and their management, organized by organ system.
Disorders of the Genital Outflow Tract and Uterus
As the cause of amenorrhea, disorders of the genital outflow tract and uterus are relatively uncommon. Congenital developmental anomalies of the genital outflow tract and uterus result from the failure of vertical fusion (imperforate hymen, transverse vaginal septum or cervical atresia) or from failure of müllerian duct development (vaginal/müllerian agenesis, AIS) and generally present at or near the expected time of menarche with primary amenorrhea. The only disorders of the genital outflow tract or uterus associated with normal genital tract anatomy are cervical stenosis and intrauterine adhesions (Asherman syndrome) or other endometrial damage resulting from surgical trauma or infection. All are acquired conditions that present as secondary amenorrhea with an onset that typically correlates closely with the time of previous insult.
Imperforate Hymen
The hymen is formed by invagination of the posterior wall of the urogenital sinus and usually ruptures spontaneously during the perinatal period. Although most cases of
imperforate hymen occur sporadically, reports of families with several affected members suggest that some cases may have a genetic and heritable cause.69
imperforate hymen occur sporadically, reports of families with several affected members suggest that some cases may have a genetic and heritable cause.69
Typically, patients with an imperforate hymen present at the expected time of menarche with complaints of cyclic perineal, pelvic or abdominal pressure or pain that results from the gradual accumulation of obstructed menstrual flow (cryptomenorrhea), and exhibit otherwise normal, symmetrical secondary sexual development for age. They also may present with acute urinary retention due to compression of the urethra and bladder by a grossly distended lower vagina.70 The genital examination reveals no obvious vaginal orifice and a thin, often bulging, blue perineal membrane at the inferior limit of a palpable, fluctuant mass (hematocolpos).
The treatment of women with an imperforate hymen centers on providing relief of symptoms related to accumulated menstrual fluid and debris. Definitive surgery should be accomplished as soon as possible because delay can lead to infertility due to inflammatory changes and to the development of severe endometriosis. Surgical correction of an imperforate hymen is straightforward. The classical procedure is to make a simple cruciate incision in the hymen to the base of the hymeneal ring and to excise its central portion to allow drainage of sequestered menstrual fluid and subsequent normal menstruation. Alternatively, to avoid any risk of damage to the hymeneal ring (a sign of virginity important to some individuals and in some cultures), a sterile puncture can be made in the center of the distended membrane and enlarged to approximately 0.5 cm in diameter to allow insertion of a 16F Foley catheter. After thorough drainage of the vagina via irrigation with sterile saline, the catheter is left in place for approximately 2 weeks to allow further drainage from the vagina and upper genital tract. A single dose of prophylactic antibiotics is prudent and estrogen cream applied locally to the hymeneal ring helps to encourage re-epithelialization.71
Transverse Vaginal Septum/Cervical Atresia
A transverse vaginal septum results when the vaginal plate, formed from the fused sinovaginal bulbs, fails to break down or canalize during embryogenesis. As could be expected, girls with a transverse vaginal septum or cervical atresia, like those with an imperforate hymen, generally present at or soon after the age of expected menarche with complaints of cyclic pelvic or abdominal pain due to obstructed menses and exhibit symmetrical, age-appropriate secondary sexual development. Physical examination reveals a normal vaginal orifice, a shortened vagina of varying length, no visible cervix, and a palpable hematocolpos in the proximal vaginal segment above the obstruction and/or a pelvic mass resulting from hematometra and hematosalpinges. A Valsalva maneuver will cause distention at the intoitus in those with an imperforate hymen, but not in those with a transverse vaginal septum or cervical atresia, and can help to distinguish the two. Imaging is necessary to define the anatomy of the disorder but laboratory investigation generally is not required. Pelvic ultrasonography can reveal the level and extent of the hematocolpos and any associated hematometra or hematosalpinges. However, abdominal/pelvic MRI provides greater anatomical detail and is recommended to more clearly define the length of the atretic segment between the lower and upper vaginas,72,73 information that is essential to planning surgical treatment. The temptation to insert a needle for diagnostic purposes must be resisted to avoid the risk of converting a hematocolpos into a pyocolpos. In rare instances, laparoscopy may be required to clarify the anatomy of the developmental anomaly. Transverse vaginal septum and cervical atresia may be accompanied by abnormalities of the upper reproductive tract, such as absent segments or atresia of the fallopian tubes or unilateral absence of the fallopian tube and ovary.74 Unfortunately, chronic retrograde menstruation frequently results in pelvic endometriosis and adhesions, which can be severe.
In all instances, every effort should be made to incise and drain the sequestered menstrual fluid from below, at the level of the obstruction. Even in complicated circumstances, continuity of the lower genital tract usually can be achieved successfully. Operative excision of painful masses from above risks damage to the bladder, ureters, and rectum and unnecessarily removes distended but otherwise healthy reproductive organs. The surgical management of a transverse vaginal septum can be challenging, and because they also are infrequently encountered, often requires consultation with specialists having the necessary training and experience. Simply described, the procedure involves excision of the septum or dissection of the atretic segment and primary anastamosis of the margins of the lower and upper vaginal canals over the site of the defect. Atretic segments of greater length may require application of a graft to bridge the gap between the lower and uppper vaginas. Because septa that appear relatively thin by physical examination and MRI may be significantly larger after decompression of the proximal hematocolpos, preparations for surgery should consider the possibility that a graft may be required.
The best surgical management for rare women with cervical atresia is controversial. Ideally, the goal would be to create a functional vagina and to preserve the uterus and fertility, but experience has proven that such heroic efforts may be associated with serious postoperative complications such as peritonitis and sepsis, recurrent obstruction, and persistent infertility, prompting many to view hysterectomy as the best management option. However, conservative surgical treatment is reasonable to consider in selected individuals. The best candidates are those recognized early, before they develop severe pelvic endometriosis and adhesions, having a well-developed lower vagina,75 although successful reconstruction and pregnancy can be achieved even in those who also require vaginoplasty.76,77
Müllerian Agenesis (Mayer-Rokitansky-Küster-Hauser Syndrome)
The failure of müllerian development is a relatively common cause of primary amenorrhea, much more frequently encountered than AIS and second only to gonadal dysgenesis in prevalence;78 in Finland, the incidence is approximately 1 in 5,000 newborn girls.79 The cause is unknown. Although usually sporadic, some cases of müllerian agensis are associated with chromosomal translocations or occur in familial aggregates, suggesting a genetic basis for the disorder. Logically, müllerian agenesis might be attributed to an activating mutation in the gene encoding antimüllerian hormone (AMH) or its receptor, causing excess AMH activity. Inactivating mutations in these genes causing persistence of müllerian structures in otherwise normally virilized males have been described.80,81 However, no activating mutations have been identified in patients with mullerian agenesis.82 The prevalence of a mutation in the galactose-1-phosphate uridyl transferase (GALT) gene (different from that associated with classical galactosemia) is increased in daughters with müllerian agenesis and their mothers.83 The observation suggests that errors in fetal or maternal galactose metabolism resulting in increased intrauterine galactose exposure may have adverse effects on müllerian development, consistent with studies in rodents wherein a high galactose diet during pregnancy delayed vaginal opening in female offspring.84 Given the relationship between classical galactosemia and premature ovarian failure, patients with müllerian agenesis who carry such a variant GALT gene mutation may be at increased risk for the same.
Patients with müllerian agenesis typically present in late adolescence or as young adults, well after menarche was expected, with primary amenorrhea as their only complaint. They exhibit normal, symmetrical breast and pubic hair development, no visible vagina, and have no symptoms or signs of crytomenorrhea because the rudimentary uteri contain no functional endometrium. However, in approximately 10%, functional islands of endometrium may result in a hematometra and symptoms of cyclic pain.85,86 Two forms
of the disorder have been described. Type A is characterized by symmetrical, muscular, rudimentary uteri, and normal fallopian tubes, and Type B by asymmetrical rudimentary uteri and absent or hypoplastic fallopian tubes.87 In the great majority of patients with müllerian agenesis, the ovaries are entirely normal, but one or both also may be undescended, hypoplastic, or associated with an inguinal hernia. Urologic anomalies are relatively common (15-40%), particularly in Type B müllerian agenesis, and include unilateral renal agenesis, ectopic or horseshoe kidney, and duplication of the collecting system(s).85,86 Skeletal malformations involving the vertebrae, the ribs, or the pelvis are observed in 10-15% of patients; some of the more common abnormalities include hemivetebrae leading to scoliosis and the Klippel-Feil syndrome, characterized by a short neck, low hairline, limited range of motion, and sometimes pain and neurologic symptoms, all relating to one or more fused cervical vertebrae.
of the disorder have been described. Type A is characterized by symmetrical, muscular, rudimentary uteri, and normal fallopian tubes, and Type B by asymmetrical rudimentary uteri and absent or hypoplastic fallopian tubes.87 In the great majority of patients with müllerian agenesis, the ovaries are entirely normal, but one or both also may be undescended, hypoplastic, or associated with an inguinal hernia. Urologic anomalies are relatively common (15-40%), particularly in Type B müllerian agenesis, and include unilateral renal agenesis, ectopic or horseshoe kidney, and duplication of the collecting system(s).85,86 Skeletal malformations involving the vertebrae, the ribs, or the pelvis are observed in 10-15% of patients; some of the more common abnormalities include hemivetebrae leading to scoliosis and the Klippel-Feil syndrome, characterized by a short neck, low hairline, limited range of motion, and sometimes pain and neurologic symptoms, all relating to one or more fused cervical vertebrae.
Although müllerian agenesis usually can be diagnosed by medical history and physical examination alone, additional evaluation is warranted to establish the diagnosis and to identify any of the urologic (renal ultrasonography) and skeletal anomalies (spinal X-rays) associated with the disorder. After puberty, a serum testosterone concentration in the normal female range effectively excludes AIS (discussed below). However, because patients with müllerian agenesis can exhibit characteristics similar to those observed in some types of male pseudohermaphroditism, a karyotype is justified and definitive. When examination raises suspicion that a uterine structure may be present, imaging is indicated. Ultrasonography may help to define the size and symmetry of any pelvic reproductive organs, but MRI is more accurate and is indicated when doubt remains.88,89 Laparoscopy usually is not necessary for diagnosis of müllerian agenesis. Although imaging frequently does not agree completely with surgical observations, detailed knowledge of the pelvic anatomy is not often needed.90 Surgery generally is indicated only in those with symptoms relating to hematometra, endometriosis, or a hernia into the inguinal canal.
The primary goal of treatment in women with müllerian agenesis—creation of a functional vagina—can be accomplished with a variety of methods, when the time is appropriate. In the large majority of cases, progressive vaginal dilation as originally described by Frank91 and later by others,92 is an appropriate and effective first choice. In motivated patients, the technique is highly successful and can create a functional vagina within 3 to 6 months.93 The procedure involves applying pressure to the point of moderate discomfort for an interval of 20-30 minutes daily, using commercially available vaginal dilators. Initially, pressure is directed posteriorly, to create a shallow pouch. After approximately 2 weeks, pressure shifts to the usual axis of the vagina. After the desired depth is achieved, dilators of increasing diameter will expand the vagina to a functional size. A variation on the technique uses a tight-fitting garment to hold the dilator in place, maintaining pressure by leaning forward on a bicycle seat mounted on a stool, or even on a bicycle.94
Operative treatment of women with müllerian agenesis generally can be reserved for those who are unable or unwilling to dedicate themselves to a program of progressive vaginal dilation and for those in whom earnest efforts fail. The traditional McIndoe procedure for surgical creation of a neovagina involves dissection of the rectovaginal space and placement of a skin graft, held in place with a soft mold until the graft becomes established.95 Subsequently, regular intercourse or vaginal dilation must be maintained to avoid risk of fibrosis and loss of function. The alternative Vecchietti operation involved internal stretching of the vaginal dimple after surgical abdominal and vaginal dissection of the vesicorectal space.96 A modification of the Vecchietti operation, performed laparoscopically, has emerged as an attractive and effective option for surgical creation of a neovagina.97,98 The procedure employs a specially designed system including a springloaded traction device, positioned on the abdomen, connected to the tip of a dilator, positioned at the introitus, via paired threads introduced with a needle inserted through small incisions in the lower abdomen and guided beneath the peritoneum to penetrate
the introitus in the midline. Tension on the threads is adjusted to maintain traction on the dilator, which is drawn upward gradually, invaginating the tissue to produce a neovagina 7-8 cm in depth over an interval of 7-10 days. Thereafter, standard vaginal dilators are employed to further extend and expand the vagina to functional dimensions over a period of a few weeks. Evidence indicates that the procedure results in a quality of sexual experience comparable to that in a sample of healthy, age-matched women of equivalent cultural and social status.98
the introitus in the midline. Tension on the threads is adjusted to maintain traction on the dilator, which is drawn upward gradually, invaginating the tissue to produce a neovagina 7-8 cm in depth over an interval of 7-10 days. Thereafter, standard vaginal dilators are employed to further extend and expand the vagina to functional dimensions over a period of a few weeks. Evidence indicates that the procedure results in a quality of sexual experience comparable to that in a sample of healthy, age-matched women of equivalent cultural and social status.98
Reassurance and support are important elements of the management of vaginal/müllerian agenesis. Affected women should be counseled that although they are infertile, normal sexual function can be expected and that genetic offspring can be achieved by in vitro fertilization (IVF) using oocytes retrieved from their own normal ovaries and the sperm of their chosen partner, with subsequent transfer of embryos to a gestational surrogate.99,100 An analysis of 34 live births resulting from IVF in 58 women with müllerian agenesis revealed no evidence to suggest a dominant pattern of inheritance and demonstrated that IVF, combined with gestational surrogacy, is a realstic option for patients with the disorder.100,101
Androgen Insensitivity Syndrome
Complete AIS (testicular feminization) is a form of male pseudohermaphroditism, the term referring to the gonadal sex (male) and the contrasting phenotype (female). The disorder, discussed in greater detail in Chapter 9 as a cause of abnormal sexual development, is the third most common cause of primary amenorrhea, after gonadal dysgenesis and müllerian agenesis. Patients with AIS have a normal male karyotype (46,XY) and testes that produce both testosterone and AMH. However, an inactivating mutation in the gene encoding the intracellular androgen receptor (located on the long arm of the X chromosome, Xq) results in an end organ insensitivity to androgen actions that prevents normal masculinization of the internal and external genitalia during embryonic development. Consequently, the external genitalia are those of a female (absent androgen action), the cervix and uterus are absent (due to normal AMH action), and the vagina is short and ends blindly (derived only from the urogenital sinus).
Patients with complete AIS appear normal at birth. Growth and development during childhood also are generally normal, although overall height usually is above average and the body habitus somewhat eunuchoid (long arms, large hands and feet). At puberty, the breasts develop, driven by estrogen derived from the peripheral conversion of high circulating testosterone levels, unopposed by the actions of androgen. The breasts may become relatively large and have subtle abnormalities; lacking the actions of progesterone, they have little glandular tissue, small nipples and pale areolae. The labia minora usually are underdeveloped and the vagina is short and ends blindly. Pubic and axillary hair does not develop, due to the absence of androgen stimulation. The testes may be intra-abdominal, but often are partially descended; more than half of patients with complete AIS have an inguinal hernia. The testes frequently are palpable in the inguinal canals, most commonly at the level of the external inguinal ring. They generally resemble any cryptorchid testes but may be nodular. After puberty, the testes contain immature seminiferous tubules lined by immature germ cells and Sertoli cells, with no evidence of spermatogenesis.
Patients with complete AIS most commonly present after the age of puberty in late adolescence or as young adults with primary amenorrhea. They exhibit asymmetrical secondary sexual development (breast development with absent or scant pubic hair), a short vagina with no visible cervix, and have no other symptoms or complaints. They also may be recognized at birth or in childhood when they may present with an inguinal mass or hernia,
particularly when the disorder is reasonably suspected because other family members such as a sister or maternal aunt are affected. Diagnosis usually is not difficult. Patients with complete AIS generally are easily distinguished from those with müllerian agenesis by the absence of pubic and axillary hair, and from those with an imperforate hymen or transverse vaginal septum by the absence of a uterus and symptoms relating to obstructed menstrual flow. In some cases, the presence of some pubic hair, due to incomplete penetrance, can be confusing or misleading. A serum testosterone concentration easily distinguishes patients with AIS because levels are normal or modestly elevated above the range observed in normal males and well above the normal range for females. Serum LH levels also are elevated, reflecting androgen insensitivity at the hypothalamic-pituitary level. A karyotype (46,XY) firmly establishes the diagnosis.
particularly when the disorder is reasonably suspected because other family members such as a sister or maternal aunt are affected. Diagnosis usually is not difficult. Patients with complete AIS generally are easily distinguished from those with müllerian agenesis by the absence of pubic and axillary hair, and from those with an imperforate hymen or transverse vaginal septum by the absence of a uterus and symptoms relating to obstructed menstrual flow. In some cases, the presence of some pubic hair, due to incomplete penetrance, can be confusing or misleading. A serum testosterone concentration easily distinguishes patients with AIS because levels are normal or modestly elevated above the range observed in normal males and well above the normal range for females. Serum LH levels also are elevated, reflecting androgen insensitivity at the hypothalamic-pituitary level. A karyotype (46,XY) firmly establishes the diagnosis.
In rare cases of incomplete androgen insensitivity, the sensitivity to androgens is greater. Consequently, pubic hair growth may accompany breast development and the clitoris may enlarge, or a phallus even may be present.102 In other rare individuals having a deficiency of the enzyme 17b-hydroxysteroid dehydrogenase (type 3), which catalyzes the conversion of androstenedione to testosterone in testicular Leydig cells, the clinical presentation may be similar, but due to impaired testosterone production rather than abnormalities in the androgen receptor. When necessary, the two disorders can be differentiated by molecular analysis of the genes encoding the androgen receptor (AR) and the enzyme (17HSDB3).103,104
The treatment of patients with complete AIS has two major components, one focusing on creation of a functional vagina, and another relating to the risk for developing malignancy in the cryptorchid testes. In patients with AIS, the options for creation of a neovagina are the same as in those with müllerian agenesis—progressive vaginal dilation and vaginoplasty. The short but distinct vagina observed in most patients speeds the progress of efforts at vaginal dilation. Good results also can be expected with surgical treatment, when necessary.105 Gonadectomy is indicated because the incidence of neoplasia in cryptorchid testes is relatively high. In one early series of 50 cases, 11 malignancies, 15 adenomas, and 10 benign cysts were observed: a 22% incidence of malignancy and a 52% overall incidence of neoplasia.106 More recent series suggest a lower 5-10% overall incidence of gonadal tumors.50,102,107,108 Whereas gonadectomy is recommended at time of diagnosis in other intersex states such as XY gonadal dysgenesis (Swyer syndrome), it is better delayed in those with AIS, for two reasons. First, the smooth pubertal development that results from endogenous hormone production is difficult to achieve with exogenous hormone treatment, and second, gonadal tumors develop less often in patients with AIS and rarely before puberty. Therefore, gonadectomy and hormone therapy (physiologic estrogen treatment) generally are best postponed until after pubertal development is complete, by approximately age 16-18. Complete AIS is the only exception to the rule that gonads with a Y chromosome should be removed as soon as a diagnosis is made. Gonadectomy usually can be accomplished endoscopically with relative ease when the testes reside within the abdomen, and via inguinal incisions when they are partially descended.109,110 In patients with the incomplete form of AIS, surgery should not be postponed because prompt gonadectomy will prevent further unwanted virilization.
In years now past, conventional wisdom warned against unthinking and “needless” disclosure of the true gonadal and chromosomal sex to patients with complete AIS for fear of undermining gender identity, but that attitude has changed. Although infertile, patients with AIS have a completely female gender identity that should be reinforced, not questioned. We strongly advocate combining a truthful education with appropriate psychological counseling for both patient and parents. Patients want, deserve, and appreciate a fuller understanding of themselves and the disorder. Moreover, because the public now has ready access to sophisticated medical information, secrecy also is no longer a practical possibility. One excellent resource is the Androgen Insensitivity Syndrome Support Group, headquartered in the United Kingdom (http://www.aissg.org/).
Cervical Stenosis
Severe cervical stenosis with complete outflow obstruction is a rare complication of cervical conization procedures or other surgical treatments for cervical intraepithelial neoplasia. When cervical stenosis causes symptoms, worsening dysmenorrhea or prolonged light staining or spotting after menses are the most common complaints; amenorrhea is a rare occurrence. Compared to other methods for limiting blood loss during conization, elective suturing appears to increase the risk for subsequent cervical stenosis and amenorrhea.111 In women with amenorrhea who have had a previous conization or other cervical surgery or ablative treatment, simple uterine sounding will establish the diagnosis of cervical stenosis and transvaginal ultrasonography will reveal any associated hematometra. The treatment for cervical stenosis is careful dilation, ideally performed under ultrasound guidance. Temporary placement of a urinary or specialized balloon catheter for an interval of approximately 2 weeks provides ongoing drainage of the uterine cavity and may help to prevent recurrence.112
Asherman Syndrome (Intrauterine Adhesions)
Asherman syndrome, first described by Joseph Asherman in 1948 and called “amenorrhoea traumatica,”113 results from intrauterine adhesions that obstruct or obliterate the uterine cavity, as a consequence of trauma. Risk for developing intrauterine adhesions is increased by inflammation, as may result from endometritis or retained products of conception, and when the endometrium is relatively thin and inactive, as it is during the postpartum period. Consequently, most cases arise in close temporal proximity to a pregnancy and are associated with surgical trauma, primarily curettage.114 In the original series of 29 cases described by Asherman, 11 had a previous postpartum hemorrhage, 15 a spontaneous abortion, 2 an elective termination, and 1 had a hydatidiform mole.113 Asherman syndrome is an uncommon but recognized complication of cesarean section, abdominal or hysteroscopic myomectomy or metroplasy, and uterine artery embolization,115 Elective endometrial ablation procedures for the management of menorrhagia frequently result in amenorrhea, by intent, but most do not have intrauterine adhesions. End organ endometrial damage and adhesions also may result from intrauterine infections such as tuberculosis and schistosomiasis, which are rare in the United States but not in other regions of the world.116,117
Although Asherman syndrome from any cause may result in amenorrhea, most women with intrauterine adhesions present with dysmenorrhea, hypomenorrhea, infertility, or recurrent pregnancy loss, rather than amenorrhea. The diagnosis of Asherman syndrome is based primarily on a high index of suspicion, based on history. In women whose history suggests the possibility, scant or no withdrawal bleeding after sequential treatment with exogenous estrogen (e.g., conjugated equine estrogens 1.25 mg daily for 21 days) and progestin (e.g., medroxyprogesterone acetate 10 mg daily for the last 5-7 days) can demonstrate end organ endometrial failure and corroborate the clinical suspicion.
However, some form of imaging ultimately is required to establish the diagnosis. Transvaginal or transabdominal ultrasonography may reveal a hematometra, but such findings are surprisingly rare. Sonohysterography or hysterosalpingography (HSG) provide more specific information regarding the location and extent of adhesions that partially or completely obliterate or obstruct the endometrial cavity or the cervical canal,118 and hysteroscopy is definitive. A variety of classification schemes have been proposed to describe the extent of intrauterine adhesions and to predict treatment outcomes,119,120,121,122,123 and 124 but none has been validated. Diagnosis of genital tuberculosis is made by endometrial biopsy (histopathology or culture) or with a nucleic acid-based test performed on an endometrial aspirate. Diagnosis of shistosomiasis is made by identifying the eggs of the parasite in urine, feces, rectal scrapings, menstrual discharge, or the endometrium.
Operative hysteroscopy is the primary method for treatment of intrauterine adhesions that may be lysed by scissors, electrodissection, or with a laser; most prefer sharp dissection, which may have less risk for causing further injury. Simultaneous laparoscopy or transabdominal ultrasonography provides useful guidance when dense scar tissue makes it difficult to enter the uterine cavity and can help to maintain orientation in a grossly distorted cavity, reducing the risk of uterine perforation. Most now advocate insertion of an intrauterine balloon catheter (left in place for approximately 7-10 days) after adhesiolysis to keep the walls of the uterine cavity separated during healing and decrease the risk of recurrence.125 Treatment with a broad-spectrum antibiotic (e.g., doxycycline 100 mg twice daily) and a non-steroidal antiinflammatory drug help to minimize risk of infection and uterine cramping while the catheter remains in place. High dose exogenous estrogen treatment (2.5 mg conjugated equine estrogens 2-3 times daily, or its equivalent) for approximately 4 weeks after surgery generally is recommended to encourage rapid endometrial re-epithelialization and proliferation; treatment with a progestin during the final week, if followed by menses, demonstrates a return of function. Despite these measures, recurrence rates are relatively high, ranging from 20% to over 60% in severe cases,126 and repeated procedures often are required to restore a normal uterine cavity.120 The surgical outcome can be assessed by postoperative HSG or by early “second-look” office hysteroscopy, which also provides the means to lyse any early recurring adhesions when still filmy.114 Menstrual function can be restored in most cases (52-88%),126 and among infertile women, live birth rates after hysteroscopic adhesiolysis generally have ranged between 25 and 35%.127 As might be expected, outcomes tend to correlate with the severity of adhesions.119,120,126,128 In those who do achieve pregnancy, the risks of preterm labor, placenta accreta, placenta previa, and postpartum hemorrhage are increased.114
Disorders of the Ovary
Disorders of the ovary include the most common causes of amenorrhea and can present as primary or secondary amenorrhea. Those resulting only from chronic anovulation and relating to PCOS, obesity, and thyroid or mild prolactin disorders were discussed earlier, in the section of this chapter devoted to the evaluation of ovarian function. The focus here is on specific disorders that result in ovarian failure and their management.
Ovarian failure occurs when few or no follicles remain that are capable of producing estradiol in response to pituitary gonadotropin stimulation. Follicular depletion may occur during embryonic life with no follicles remaining by infancy or early childhood, after puberty has begun but before menarche, or at some later time before menopause normally would be expected. Consequently, depending on when the available supply of ovarian follicles is functionally depleted, puberty may not occur, it may begin normally but stop before the first menses, or it may progress normally to and beyond menarche with secondary amenorrhea having onset at some later point in time.
In a few women with rare genetic disorders, hypergonadotropic hypogonadism results from a functional ovarian failure due to abnormalities of follicular development, rather than from follicular depletion.
Gonadal Dysgenesis
Gonadal dysgenesis is defined as an incomplete or defective formation of the gonads, resulting from a disturbance in germ cell migration or organization, caused by structural or numerial sex chromosome abnormalities or mutations in the genes involved in formation of
the urogenital ridge and sexual differentiation of the bipotential gonad. Gonadal dysgenesis is among the most common causes of primary amenorrhea (approximately 30-40%). Due to the absence of ovarian follicles or their accelerated depletion during embryogenesis or the first few years of life, the gonads contain only stroma and appear as fibrous streaks. The large majority of patients with gonadal dysgenesis have an obvious abnormality involving an X chromosome. Aproximately 25% of affected individuals have a normal 46,XX karyotype and may harbor a more subtle abnormality invoving one or more specific genes on the X chromosome that are required for normal ovarian function; some with 46,XX gonadal dysgenesis also have neurosensory deafness, the combination known as Perrault syndrome. By far, the most common form of gonadal dysgenesis is Turner syndrome.
the urogenital ridge and sexual differentiation of the bipotential gonad. Gonadal dysgenesis is among the most common causes of primary amenorrhea (approximately 30-40%). Due to the absence of ovarian follicles or their accelerated depletion during embryogenesis or the first few years of life, the gonads contain only stroma and appear as fibrous streaks. The large majority of patients with gonadal dysgenesis have an obvious abnormality involving an X chromosome. Aproximately 25% of affected individuals have a normal 46,XX karyotype and may harbor a more subtle abnormality invoving one or more specific genes on the X chromosome that are required for normal ovarian function; some with 46,XX gonadal dysgenesis also have neurosensory deafness, the combination known as Perrault syndrome. By far, the most common form of gonadal dysgenesis is Turner syndrome.
Turner Syndrome
Turner syndrome is a well known and thoroughly studied disorder, classically associated with a 45,X karyotype, but also with an assortment of other structural X chromosome abnormalities (deletions, ring and iso-chromosomes), any of which may be present in all or only in some of the cells of the body (mosaicism), depending on the stage of embryonic development at the time they arise. The disorder is discussed thoroughly in Chapter 9, as a cause of abnormal sexual development, and is more briefly summaried here.
The classical phenotype of Turner syndrome includes short stature, absent sexual development, a webbed neck, low set ears and posterior hairline, widely-spaced nipples (“shield chest”) short fourth metacarpals, and an increased carrying angle at the elbow (“cubitus valgus”). Evidence indicates that the specific phenotype of patients with Turner syndrome relates, in part, to the parental origin of their X chromosome; most with a 45,X karyotype retain the maternal X.129
If not recognized by phenotype or poor growth during childhood, patients with Turner syndrome generally present at or near the time of expected puberty with primary amenorrhea and absent secondary sexual development. The diagnosis of Turner syndrome generally can be made easily, based on the phenotype and findings of hypergonadotropic hypogonadism. A karyotype is definitive, and specifically indicated, in part because it may reveal a cell line containing a Y chromosome otherwise not suspected or identified (e.g., 45,X/46,XY); approximately 5% of women with Turner syndrome have a karyotype containing all or part of a Y chromosome.130 Futher analysis with fluorescence in situ hybridization (FISH) using one or more probes specific for segments of the Y chromosome will identify another 5% having occult Y chromosome material.130,131 Whereas it is important to identify a Y chromosome because affected individuals are at significant increased risk for developing gonadoblastoma (20-30%), that risk appears lower (5-10%) in women with Turner syndrome, and limited to those having detectable Y chromosome on their karyotype. FISH analysis is most clearly indicated for those exhibiting any evidence of virilization or having a chromosomal fragment of uncertain origin (discussed in chapter 9).132
Mosaicism in women with Turner syndrome has important clinical implications besides those relating to a cell line containing a Y chromosome. In those with a mosaic 46,XX cell line (e.g., 45,X/46,XX), the gonad may contain functional ovarian cortical tissue, resulting in some degree of sexual development, or even menses and the possibility of pregnancy. Approximately 15% of patients with Turner syndrome begin but do not complete pubertal development and approximately 5% complete puberty and begin menstruation.133 As might be expected, the phenotype varies, with some appearing normal and attaining normal stature before experiencing ovarian failure when the limited supply of follicles is exhausted. Natural pregnancies do occur in women with Turner syndrome, but they are rare
and associated with a relatively high risk for sex chromosome aneuploidy and spontaneous abortion.
and associated with a relatively high risk for sex chromosome aneuploidy and spontaneous abortion.
Women with Turner syndrome may have a wide variety of medical problems having health implications as or more important than those relating to or resulting directly from hypogonadism.132 Approximately one-third has cardiovascular anomalies, including a bicuspid aortic valve, coarctation of the aorta, mitral valve prolapse, and aortic aneurysm. Renal anomalies also are common and include horsehoe kidney, unilateral renal agenesis or pelvic kidney, rotational abnormalities, and partial or complete duplication of the collecting system(s). Autoimmune disorders are common in Turner syndrome and include thyroiditis, type 1 diabetes, autoimmune hepatitis and thrombocytopenia, and celiac disase. Hearing loss also is common.134,135 Consequently, additional and periodic medical evaluation is indicated and should include the following132:
Echocardiography (at diagnosis, at least once between the ages of 12 and 15 years, and every 5 years if normal; more often if abnormal);
Renal ultrasonography (once if normal, every 3-5 years if abnormal);
TSH and free T4 (at diagnosis and every 1-2 years);
Complete blood count, fasting glucose, lipid profile, renal function tests, and liver enzymes (every 2 years);
Anti-endomysial antibodies, to detect celiac disease (at diagnosis);
Audiometry (at diagnosis, at least once during the teen years or young adulthood, and every 10 years if normal).
Average intellectual performance is within the normal range,136 although the prevalence of attention-deficit/hyperactivity disorder (ADHD) is increased in girls with Turner syndrome.137 Overall mortality is increased approximately 3-fold and relates primarily to circulatory disease (e.g., hypertension), diabetes, liver and renal disease.138 Overall cancer risks in women with Turner syndrome are similar to those in the general population, but the incidence of CNS tumors, bladder cancer, and endometrial cancer may be increased, and the risk of breast cancer is decreased.139 With early diagnosis and treatment with growth hormone (GH), a final height greater than 150 cm (59 inches) can be achieved in most patients with Turner syndrome. The most important determinants of final height are the dose of GH and the duration of treatment before estrogen therapy begins. Treatment with GH generally should begin as soon as height falls below the fifth percentile of normal female growth and must be individualized, according to response.132
Treatment with estrogen must be timed carefully, with the goals of minimizing its adverse effects on growth and adult height and inducing puberty at an approximately normal age. Ideally, estrogen therapy should begin no later than age 15 and not before age 12 when growth is a priority, unless height already has been maximized. Estrogen therapy should begin at a low dose (e.g., 0.25-0.5 mg micronized estradiol or its equivalent), increasing gradually at intervals of 3-6 months according to response (Tanner stage, bone age), with the goal of completing sexual maturation over a period of 2-3 years. When vaginal bleeding first occurs, or after 12-24 months of estrogen therapy, a progestin (e.g., medroxyprogesterone acetate) should be added to the treatment regimen to complete development, prevent dysfunctional bleeding, and to protect the endometrium from the effects of unopposed estrogen.132
Oocyte donation offers the possibility of pregnancy to patients with Turner syndrome, but the cardiovascular demands of pregnancy pose unique and potentially serious risks that must be carefully considered. The risk of death during pregnancy is increased as much as 100-fold, primarily due to complications of aortic dissection or rupture. Risk is greatest for those with preexisting abnormalities such as a bicuspid aortic valve or a dilated aortic root, but even those without such findings remain at risk. Consequently, Turner syndrome generally should be regarded as a relative contraindication to pregnancy. Those expressing
serious interest in oocyte donation must receive thorough evaluation and counseling, and those having any significant cardiac abnormality should be strongly discouraged.140
serious interest in oocyte donation must receive thorough evaluation and counseling, and those having any significant cardiac abnormality should be strongly discouraged.140
Swyer Syndrome (46,XY Gonadal Dysgenesis)
Swyer syndrome is a distinctly different and less common form of gonadal dysgenesis, characterized by a 46,XY karyotype. Despite the presence of a Y chromosome, the phenotype is female because the dysgenetic (streak) gonads produce neither AMH nor androgens. Consequently, the vagina, cervix, uterus, and fallopian tubes develop normally and the internal and external genitalia do not masculinize.141 In at least 10-15% of affected individuals, a mutation of the SRY gene (Sex-determining Region of the Y chromosome; located on the short arm, Yp11.3) is the cause.142 In the remainder, no cause can be determined, although mutations in SRY regulatory elements or in other genes involved in the testis-determining pathway have been implicated (SF1, SOX9, WT1, CMRT1).143,144 and 145
Patients with Swyer syndrome generally present after the expected time of puberty with delayed sexual maturation and primary amenorrhea. The presence of pubic hair reflects a normal adrenarche. Evaluation reveals hypergonadotropic hypogonadism, prompting a karyotype that establishes the diagnosis. Gonadectomy is indicated soon after diagnosis due the significant risk for malignant transformation in occult testicular elements (20-30%). Gonadoblastoma is a premalignant germ cell tumor, unique to intersex states like Swyer syndrome, and may contain or give rise to other highly malignant tumors, including dysgerminoma, endodermal sinus tumor, embryonal and choriocarcinomas; evidence suggests they originate from clonal expansion of surviving germ cells in areas of undifferentiated gonadal tissue.146
Patients with Swyer syndrome exhibit normal growth and intellectual development, have no increased prevalence of any specific medical problems, and require no specific monitoring or treatment beyond that relating to hormone therapy aimed at inducing sexual maturation. The same sequential sex steroid treatment regimen described above for patients with Turner syndrome can be applied successfully in those with Swyer syndrome. Pregnancy, achieved with in vitro fertilization using donor oocytes, is a realistic expectation and has not been associated with any specific risks or complications.147
46,XX Gonadal Dysgenesis
Some individuals with primary amenorrhea and gonadal dysgenesis (streak gonads) have a normal 46,XX karyotype, providing indirect evidence that autosomal genes also play a critical role in ovarian differentiation. Affected women are normal in stature and, in most cases, have no apparent somatic anomalies. A wide variety of candidate genes has been identified, primarily via experiments involving murine knock-out models, including several that encode DNA and RNA binding proteins and transcription factors expressed during oogenesis.148
Premature Ovarian Failure
Premature ovarian failure (POF), traditionally defined as hypergonadotropic hypogonadism and amenorrhea arising before the age of 40, is a heterogeneous disorder that varies widely in cause and phenotype. Whereas the term POF is well-entrenched in the medical literature,
an alternative term—“premature ovarian insufficiency”—has been proposed to more accurately reflect the continuum of decreased ovarian function observed in affected women,149 acknowledging that many exhibit intermittent ovarian function and ovulation and that 5-10% may conceive and deliver a pregnancy.150,151
an alternative term—“premature ovarian insufficiency”—has been proposed to more accurately reflect the continuum of decreased ovarian function observed in affected women,149 acknowledging that many exhibit intermittent ovarian function and ovulation and that 5-10% may conceive and deliver a pregnancy.150,151
Premature ovarian failure (POF) generally results in secondary amenorrhea at some time after puberty is completed, but also may occur at any time before menarche and is distinguished from gonadal dysgenesis on the basis of ovarian morphology and histology; instead of streak gonads, the ovaries more closely resemble those of postmenopausal women. Approximately 1% of women will develop POF before the age of 40 years. In a cross-sectional survey of women aged 40-55 conducted at seven sites in the United States to determine eligibility for a community-based, multi-ethnic longitudinal sudy of the perimenopause (The Study of Women Across the Nation, SWAN), premature menopause was reported by 1% of Caucasians, 1.4% of African Americans and Hispanics, 0.5% of Chinese, and 0.1% of Japanese women.152
Important known causes of POF include numerical and structural chromosomal abnormalities, fragile X (FMR1) premutations, autoimmune disorders, radiation therapy, and chemotherapy. Whereas history alone can identify the latter two, the first three merit specific consideration and exclusion. In these and other rare disorders associated with ovarian failure such as galactosemia, the basic pathophysiology involves accelerated follicular atresia. In other rare genetic disorders, mutations in genes encoding intraovarian regulators, steroidogenic enzymes, gonadotropins, or their receptors result in impaired or abnormal follicular development, but the end result is much the same—functional ovarian failure. In most women with POF, a specific cause cannot be identified, but evidence implicating a number of genetic factors is growing rapidly.148
Numerical and Structural Chromosomal Abnormalities
A wide variety of numerical and structural chromosomal abnormalities may be identified in women presenting with ovarian failure. A review of karyotypes obtained in women with secondary amenorrhea revealed the spectum of possibilities and demonstrates the importance of a karyotype in women with POF.153 Half of the observed abnormalities were numerical, involving X chromosome mosaicism (including 45,X, 46,XX, and 47,XXX cell lines) or Y chromosome mosaicism (including 46,XY, 47,XYY, and 47,XXY cell lines); the remainder included an assortment of X chromosome translocations, deletions and other structural abnormalities, and even some with a pure 46,XY karyotype.
Chromosomal deletions and translocations involving either the short (Xp) or the long arm (Xq) of the X chromosome may be identified in women with POF. Only about half of those with deletions involving the short arm of the X chromosome present with primary amenorrhea and gonadal dysgenesis; the remainder menstruates and often presents with POF. Presumably, the short arm of the X chromosome contains genes essential for ovarian germ cell function. Although the specific genes involved are unknown, likely candidates include BMP15 (Bone Morphogenic Protein) and other members of the TGFb (Transforming Growth Factor) superfamily.148 The long arm of the X chromosome also contains genes crucial for normal ovarian function. Likely candidate genes, identified by the phenotypes associated with deletions and translocations involving Xq, include XIST (the X- Inactivation gene), DACH2 (encoding a transcription factor), and QM (encoding a ribosomal protein).148,154
Fragile X (FMR1) Premutations
A spectrum of clinically important disorders, including POF, involves a dynamic trinucleotide (CGG) repeat sequence mutation in the X-linked FMR1 gene, located near
the terminal end of the long arm of the X chromosome (Xq27.3). The normal FMR1 gene contains approximately 30 repeats. The fully expanded form of the mutation, characterized by more 200 CGG repeats, results in fragile X syndrome (FXS), the most common known genetic cause of mental retardation and autism. The premutation, characterized by 55-200 repeats, is associated with two disorders distinct from FXS. One is the fragile X-associated tremor/ataxia syndrome (FXTAS), a neurologic disorder that affects males primarily, as might be expected for an X-linked disorder. The other is POF, affecting approximately 15% of women who carry the premutation.56
the terminal end of the long arm of the X chromosome (Xq27.3). The normal FMR1 gene contains approximately 30 repeats. The fully expanded form of the mutation, characterized by more 200 CGG repeats, results in fragile X syndrome (FXS), the most common known genetic cause of mental retardation and autism. The premutation, characterized by 55-200 repeats, is associated with two disorders distinct from FXS. One is the fragile X-associated tremor/ataxia syndrome (FXTAS), a neurologic disorder that affects males primarily, as might be expected for an X-linked disorder. The other is POF, affecting approximately 15% of women who carry the premutation.56
In the full mutation, the expanded number of trinucleotide repeats in the 5′ untranslated region of the gene results in hypermethylation that extends into the promoter region and silences the gene. Consequently, there is little or no production of mRNA and its product, the fragile X mental retardation protein (FMRP), which is an RNA binding protein that functions as a translational suppressor. Ultimately, the result is an overexpression of mRNAs, leading to the clinical features of FXS.155 The incidence of FXS is approximately 1 in 4,000 males and 1 in 4,000-8,000 females.56 Although affected females are protected to some extent from the full impact of the mutation, due to X-inactivation, approximately 70% have a borderline or lower IQ or other functional deficits.156,157 and 158
In the premutation, the trinucleotide repeat sequence is not methylated, the gene functions, and FMRP is produced. However, the condition is quite different from the usual unaffected carrier state in two important ways: (1) premutation carriers are at risk for developing disorders different from FXS, and (2) the affected alleles are unstable and at risk for expansion from the premutation to the full mutation. FXTAS is a progressive neurodegenerative disorder affecting male premutation carriers, generally after age 50, causing intention tremor, ataxia, autonomic dysfunction, cognitive deficits, behavioral abnormalities, and peripheral neuopathy.159 Female premutation carriers may develop the disorder, but do so infrequently; unfavorable X chromosome inactivation may increase the risk of FXTAS in women.160 However, women with a FMR1 premutation frequently develop POF. The prevalence of premutations is approximately 15% among women with familial POF and lower, but still significant (1-7%), in those having no family history of POF.56 The variation in prevalence may be explained by the relationship between the probability of POF and the size of the CGG repeat sequence; risk for POF increases with the number of repeats, between 59 and 99, but rises no further and even decreases for those with more than 100.161,162 Women with a modestly expanded repeat sequence (41-58 repeats) also may be at increased risk for POF.163,164
Women who carry the FMR1 premutation often exhibit signs of early reproductive aging. The lengths of their cycle and follicular phase are shorter, FSH levels across all phases of the cycle are higher, and inhibin levels are lower, compared to those in normal women.165 They also enter menopause approximately 5 years earlier than average.161 Exactly why or how the FMR1 premutation predisposes to POF is not entirely clear but may involve gain-of-function toxicity, due to overexpression of mRNAs, resulting in accelerated follicular atresia.
The inheritance of FMR1 mutations and premutations follows the basic pattern of an X-linked disorder; females transmit the abnormality to 50% of their offspring and male carriers to all of their daughters and none of their sons. The pattern of inheritance is complicated by the meiotic instability of the trinucleotide repeat sequence, which has a tendency to expand when passed from the mother to her progeny, thereby increasing the risk for FXS with each generation, a phenomenon known as “anticipation.” The risk for expansion depends on the size of premutation; a repeat sequence numbering between 59 and 79 expands to the full mutation less than half the time, but one larger than 90 does so more than 90% of the time.166 In contrast, the size of the premutation remains relatively stable when transmitted from fathers to their daughters and rarely expands to the full mutation,167 possibily because large repeat sequences are highly unstable in developing sperm and only
smaller premutations can be transmitted. Intermediate or “gray zone” repeat sequences numbering between 45 and 54 may expand to premutation size across generations. Due to the complexity of the inheritance of FMR1 premutations, all carriers should receive formal genetic counseling. Issues that must be addressed include the risk for infertility and early menopause, the risk for and implications of transmitting the premutation, and the possibility that other family members may be affected, in a variety of ways, raising still other and obvious ethical concerns.
smaller premutations can be transmitted. Intermediate or “gray zone” repeat sequences numbering between 45 and 54 may expand to premutation size across generations. Due to the complexity of the inheritance of FMR1 premutations, all carriers should receive formal genetic counseling. Issues that must be addressed include the risk for infertility and early menopause, the risk for and implications of transmitting the premutation, and the possibility that other family members may be affected, in a variety of ways, raising still other and obvious ethical concerns.
All women with POF should be offered screening for the FMR1 premutation. In sum, guidelines issued by the American College of Medical Genetics, the American College of Obstetricians and Gynecologists, and the American Society for Reproductive Medicine all recommend testing for women with unexplained POF.58,168 Whereas there is not yet a clear consensus on whether testing is indicated for all infertile women under age 40 with modestly elevated FSH levels suggesting a diminished ovarian reserve, all agree that testing should be offered to those having a family history of POF, FXS, or FXTAS or having relatives with unexplained mental retardation or autism. Arguments against more liberal or widespread population screening center on the limited resources available to provide the complicated counseling required and the uncertainties surrounding the risks associated with alleles in the intermediate range, which are common.
Up to 5-10% of women with POF who carry an FMR1 premutation will conceive after diagnosis, without medical intervention, but there is no evidence that any treatment other than oocyte donation can increase the likelihood of pregnancy.151
Autoimmune Disorders
Autoimmune disease is one of the known causes of POF, accounting for approximately 4% of cases.169 There is substantial evidence to indicate that autoimmunity is the cause of POF in women who also exhibit signs of adrenal autoimmunity.60 Autoimmune oophoritis may occur as part of a Type I or II autoimmune polyglandular syndrome (APS) associated with autoantibodies to multiple endocrine and other organs. Type I APS (also known as autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy) presents in childhood, typically as hypoparathyroidism (89%) or mucocutaneous candidiasis (75%), and often is accompanied by adrenal insufficiency (60-80%) and POF (60%); the cause is a mutation in the AIRE (autoimmune regulator) gene, located on chromosome 21. Type II APS has an adult onset and is characterized by adrenal insufficiency (100%) and thyroid autoimmunity (70%) or type 1 diabetes (50%); 25% of women with the disorder have amenorrhea and 10% have POF.59 POF also has been described in women with systemic lupus erythematosus and myasthenia gravis.
The mechanism that stimulates or causes ovarian autoimmunity is unknown, but might involve a virus or other cause of damage to ovarian tissue that renders it antigenic, or a basic failure in immune regulation resulting in a loss of tolerance to some component of ovarian tissue. The hallmark of autoimmune oophoritis is a lymphocytic infiltrate surrounding secondary and antral follicles but not primordial follicles,169,170 strongly suggesting that steroid hormone producting cells in the theca contain the inciting antigen. Observations of normal inhibin B production in otherwise hypogonadal women with autoimmune oophoritis further suggest that the theca is selectively targeted and that granulosa cells are spared.171 Almost all women with documented autoimmune oophoritis have circulating antibodies directed against steroidogenic enzymes such as 21-hyroxylase, 17a-hydroxylase, and side chain cleavage.60,169 The diagnosis of autoimmune ovarian failure therefore hinges on the demonstration of autoantibodies against steroidogenic cells. A positive test for adrenal or 21-hydroxylase antibodies is sufficient to establish a diagnosis of autoimmune ovarian failure. A commercially available serum anti-ovarian antibody test had poor predictive value and an unacceptably high false-positive rate.172 Ovarian biopsy solely for diagnosis
of autoimmune ovarian failure is unnecessary and is not recommended. Whereas antibodies against membrane-bound receptors may cause diseases such as mayasthenia gravis, the failure to detect antibodies against the FSH receptor in women with POF suggests they rarely, if ever, are the cause.173 Women with autoimmune ovarian failure may develop large luteinized follicular cysts, possibly due to increased gonadotropin stimulation in response to impaired follicle development and function.171
of autoimmune ovarian failure is unnecessary and is not recommended. Whereas antibodies against membrane-bound receptors may cause diseases such as mayasthenia gravis, the failure to detect antibodies against the FSH receptor in women with POF suggests they rarely, if ever, are the cause.173 Women with autoimmune ovarian failure may develop large luteinized follicular cysts, possibly due to increased gonadotropin stimulation in response to impaired follicle development and function.171
The strong association between autoimmune adrenal and ovarian failure justifies screening for anti-adrenal antibodies in all women with POF, at the time of diagnosis. The most sensitive and useful methods for their detection are indirect immunofluorescence assays using adrenal tissue as substrate and immunoprecipation assays for antibodies against the 21-hyroxylase enzyme (CYP21). Patients with positive anti-adrenal antibodies should be further evaluated to exclude asymptomatic adrenal insufficiency, by measuring the morning (6:00-9:00 A.M.) serum cortisol level. A value greater than 18 mg/dL effectively excludes clinical adrenal insufficiency; those with lower values require further evaluation with an ACTH stimulation test to determine whether ACTH reserve is sufficient to meet demand during times of stress. The test is performed by measuring the serum cortisol concentration before and 60 minutes after administering cosyntropin (synthetic ACTH 1-24; 0.25 mg) intramuscularly or intravenously; a stimulated cortisol concentration ≥18 μg/dL is a normal response. Those with negative tests for adrenal antibodies should be followed and monitored with repeat testing at intervals, because POF may precede onset of adrenal insufficiency by up to several years and its autoimmune cause may not, at first, be recognized.59
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


