Surgical Care of Conditions Presenting in the Newborn
Andrea T. Badillo
Nancy M. Bauman
Michael J. Boyajian
Ashanti L. Franklin
M. Taylor Fordham
Mikael Petrosyan
Anthony D. Sandler
The fragile nature and limited reserve of newborn infants are superimposed on the stress of the surgical condition and its operative correction. Temperature regulation, fluid, blood, and glucose administration, and monitoring of respiratory and cardiovascular performance are critical in managing the surgical neonate.
Venous and arterial access for fluid administration and monitoring is essential. Peripheral venous access with a 22- or 24-gauge catheter provides adequate access for the most vigorous fluid resuscitation. Central venous access may be necessary in many surgical neonates with long-term issues and is most frequently obtained by long intravenous percutaneous catheters (PIC) in the unstable newborn, or if peripheral access is unsuccessful. Umbilical vessel catheterization provides vascular access and arterial monitoring. The umbilical catheter may be maintained during most operative procedures. Peripheral arterial access may be obtained using radial, ulnar, or posterior tibial puncture or cutdown. Surgical central venous access with tunneled catheters is available if access is not obtained or long-term access is needed.
Total fluid requirements are often significantly greater than maintenance fluid requirements. Before the operation, there may be extraordinary losses from the gastrointestinal (GI) tract or inflamed peritoneum. These fluid losses continue in the postoperative period and are superimposed on the sodium and water retention associated with the metabolic stress response to surgery. Postoperative oliguria may result from a surge in antidiuretic hormone, intravascular volume depletion, or both, limiting the utility of urine volume alone to serve as a measure of adequate fluid replacement. Assessments of skin temperature, quality of peripheral pulses, serial measurements of weight, hematocrit, serum electrolytes, and osmolality supplement urine volume as indicators of adequate volume replacement (1).
Temperature maintenance is a critical concern for all newborns, but in particular for those undergoing diagnostic studies and operative procedures in radiology and operating suites. A variety of means of temperature support may be utilized, including warming the environment, heat lamps, heating blankets, scalp and extremity wrapping, heated intravenous fluids and blood products, and heated inhaled anesthetic agents and surgical irrigating fluids. Temperature must be constantly monitored and every effort made to minimize exposure to cold stress. Hypothermia is a potentially lethal condition, and the importance of temperature support is critical (2).
Anesthetic and postoperative pain management can minimize the magnitude of the stress response and accelerate the neonate’s return to normal homeostasis in terms of cortisol, catecholamine, and insulin modulation (3). Optimal neonatal outcomes require collaboration between all specialists involved in the care of the newborn.
▪ UPPER AERODIGESTIVE TRACT AND CONGENITAL CERVICAL ANAMOLIES
Aberrant fusion of midline structures arises from defects in embryogenesis occurring in the first 4 to 8 weeks of gestation and most commonly affects the lip, palate, larynx, trachea, and rarely the nose.
Cleft Lip
Clefts of the lip or palate occur in approximately 1 of every 600 to 700 Caucasian newborns. The frequency is doubled in Asians and halved in African Americans. Cleft lip occurs somewhat more often in male patients and on the left side. The defect probably results from lack of the mesodermal reinforcement of the junction of the nasomedial and lateral facial processes that normally takes place in the 6th to 7th week of gestation. Multiple genetic influences seem to be more important than environmental factors. The cleft deformity ranges from minor notching to complete separation of the entire lip and nasal floor (Fig. 41.1). The defect may involve the lip, the lip and palate, or only the palate, and it may be unilateral or bilateral. Median cleft lip is rare and is usually associated with hypotelorism, microcephaly, and early death.
Airway obstruction is not typically a consequence of isolated cleft lip or palate. Initial care focuses on feeding the infant and counseling the parents. Swallowing and airway protection should be normal, but the negative pressure of the normal suck is vented through the cleft resulting in inadequate inflow. Fatigue during feeding is common and may mimic satiation. Although suckling is not altogether discouraged, a baby with a complete cleft lip or any degree of cleft palate should be expected to suffer mechanical feeding difficulty. The solution takes the form of an enlarged nipple aperture, a compressible bottle, a chambered (Haberman) nipple, or a syringe feeder. With the use of a positive-pressure delivery system, the feeding schedule should be normal.
Lip closure is usually carried out at around 3 months of age. The major goals are muscle continuity, balanced lip height, the normal Cupid-bow lip shape, a smooth and pout-free lip margin, a good nasal sill, adequate sulcus lining, and a minimal, well-placed scar. The wide, complete unilateral and the complete bilateral clefts present greater challenges. Preliminary lip adhesion for the unilateral case or presurgical orthodontics can improve anatomic associations and facilitate the definitive surgery. Residual nasal deformity is often a stubborn problem and may require secondary surgery.
Cleft Palate
The embryologic palatal shelves initially hang vertically and then rise to meet and fuse from front to back between weeks 7 and 12 of gestation. Interference with this process may result in complete, incomplete, or submucous cleft of the palate. Initial care is discussed in the section on cleft lip.
The major significance of this defect is the effect on speech. Normal modulation of speech requires reliable, dynamic palatal separation of the mouth from the nose. This requires a palate of adequate length, suppleness, and muscle power. Velopharyngeal incompetence or incomplete nasal closure results in hypernasal speech and significant communication disability.
Chronic or recurrent effusion and infection in an otherwise normal ear is common in the child with a cleft palate because eustachian tube function is compromised. This child usually needs myringotomies and ventilation tubes.
Early surgery seems to have a negative effect on facial growth, but the trend is toward closure during infancy because of the improved speech results. Most American surgeons choose 9 to 12 months of age as optimal timing for a single-stage closure.
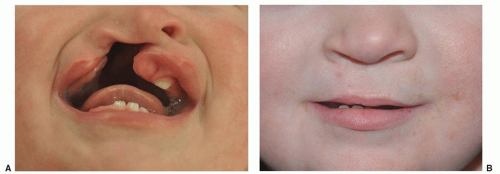 FIGURE 41.1 Complete right cleft lip (A) preoperative and (B) postoperative. |
Palatal closure is accomplished with local soft tissue. Mucoperiosteal flaps are mobilized and closed in the midline, with oral and nasal lining, effecting muscle apposition and retroposition. No bone reconstruction is involved. The goal is normal speech, and this is achieved in approximately 85% of patients. A second operation produces good results for almost all the remaining infants.
An essential concept in the treatment of these children is a multidisciplinary approach. The patient should be followed through adolescence by a team consisting of a plastic surgeon, otolaryngologist, audiologist, pedodontist, orthodontist, speech pathologist, geneticist, pediatrician, and social worker.
Pierre-Robin Sequence
The PRS is characterized by retrognathia or micrognathia (i.e., small or recessed jaw or chin), glossoptosis, airway obstruction, and cleft palate. The lack of forward support of the tongue allows it to fall back and compromise the airway. The basic defect may result from intrauterine restriction of mandibular growth.
Intensive monitoring, which may include a home apnea monitor, is necessary for many patients. The airway can usually be maintained by conservative measures. Prone positioning allows the tongue to fall forward and can be effective. A nasal airway can provide temporary relief. An appropriately apertured board may facilitate this positioning, and a nasal airway may be useful. Early gavage feedings may obviate hazardous oral feedings. A lip-tongue adhesion may be performed in more difficult cases, but its effectiveness varies. For appropriately selected candidates, mandibular osteotomies and distraction can address the issue directly. Tracheotomy placement should be avoided if possible, but it is sometimes the only safe choice. Management should be as conservative as the clinical situation permits. The airway problem is typically self-limited, resolving as the child grows.
Congenital Cervical Lesions
Congenital neonatal neck lesions are far more common than infectious or neoplastic lesions, and most arise from aberrant development of the branchial apparatus, a series of grooves, pouches, and arches that contribute to the development of the ears, lower face, and neck structures.
Thyroglossal Duct Remnants
Midline thyroglossal duct cysts most commonly present later in childhood but can appear in newborns as a midline cystic neck mass that characteristically ascends with swallowing. These anomalies are embryologic remnants of the developing thyroid gland as it descends from the foramen cecum of the tongue to the midline of the neck. The tendency for repeated infection warrants removal of thyroglossal duct cysts sometime during childhood along with the intimately associated midline segment of the hyoid bone via a Sistrunk procedure (Fig. 41.2).
Branchial Cleft Anomalies
Branchial arch anomalies are characteristically located at the anterior midlevel border of the sternocleidomastoid muscle and may present as cysts, sinus tracts, or fistulae. Surgical excision is usually indicated due to recurrent drainage and infections. Second branchial cleft cysts are most common and often present with a sinus tract extending to the skin or to the tonsillar fossa. First branchial cleft anomalies with an internal communication drain to the
middle ear or external auditory canal while third and fourth branchial arch anomalies drain to the hypopharynx or the esophagus.
middle ear or external auditory canal while third and fourth branchial arch anomalies drain to the hypopharynx or the esophagus.
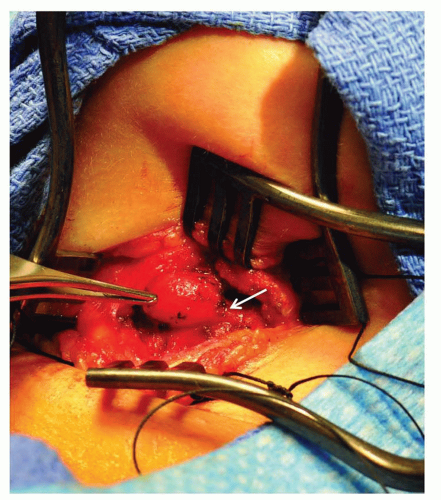 FIGURE 41.2 Operative excision of a thyroglossal duct cyst. Note the lesion’s midline location and its attachment to the underlying hyoid bone (arrow). |
Preauricular Sinuses, Skin Tags, and Cartilaginous Remnants
Preauricular skin tags and preauricular sinuses arise due to abnormal formation of the ear hillocks during the 6th week of gestation. Infants with preauricular pits have a slightly higher risk of sensorineural hearing loss (4). Preauricular pits are usually isolated but if hearing loss is also present, genetic testing for EYA1 and SIX1 is indicated to exclude branchiootic or branchiootorenal syndrome. Unsightly skin tags and cartilage rests may warrant removal for cosmetic reasons. For pedunculated lesions, this can be completed by placing a constricting suture around the narrow base in the newborn nursery, much to the delight of the parents. Preauricular sinuses are benign, blind-ending pouches that are only removed during childhood, if they repeatedly become infected.
Cervical Vascular Anomalies
Lymphatic malformations are the most common cervical vascular anomalies seen in the newborn period, although cervical hemangiomas, arteriovenous, and venous malformations may also occur during this time. Eighty percent of lymphatic malformations arise in the neck and represent sequestered lymphatic channels that fail to communicate with larger lymphatics or draining veins (Fig. 41.3). Nearly half of LMs are evident at birth with most of the remaining lesions appearing during the first decade of life. These soft, cystic congenital anomalies are classified as macrocystic, microcystic, or mixed lesions and characteristically insinuate themselves around critical vessels and nerves. LMs may partially or completely shrink, but this appears to be limited to macrocystic lesions and occurs in fewer than 15% of cases. Conversely, LMs can undergo sudden and rapid enlargement from internal bleeding or infection that can progress to sepsis. Massive LMs of the upper aerodigestive tract can cause airway obstruction necessitating tracheotomy tube placement. Fortunately, most LMs of infancy are asymptomatic, cosmetically deforming lesions for which treatment can usually be deferred until after 6 months of life. Macrocystic lesions are most amenable to sclerotherapy under general anesthesia; and OK432, doxycycline, and ethanol are the most common agents used (5). Bleomycin has recently shown some efficacy for microcystic lesions, although most require surgical excision with an attendant risk of neuropraxia and recurrence even in the most fastidious of hands. Infants with massive LMs require multiple surgical and medical interventions over the course of many years to achieve a favorable outcome. Sirolimus, an mTOR pathway inhibitor, may prove a promising treatment for select patients with massive LMs, but additional trials are warranted (6).
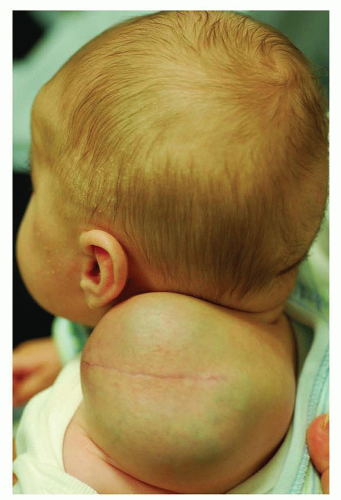 FIGURE 41.3 Classic macrocystic lymphatic malformation. While the cervical mass is large, there is no urgency to intervene if the lesion does not interfere with respiration and if the patient is otherwise asymptomatic. |
Upper Airway Obstruction Causing Respiratory Distress
Evaluation of the newborn in respiratory distress requires prompt assessment of the severity of the problem and its potential etiology(ies). Nasal flaring, tachypnea, use of accessory muscles of respiration, and suprasternal retractions are all signs of respiratory distress. The presence of stertor or stridor indicates anatomic obstruction; however, in advanced cases of obstruction, the breathing may be deceivingly quiet as the respiratory effort and air movement decline. In obstructive lesions, CO2 levels rise before oxygen levels decline, and by the time hypoxemia occurs the airway obstruction may be markedly advanced.
Precise assessment of the respiratory sounds helps to determine the site of the obstruction. Stertor is a low-pitched “snorty” sound that arises from nasal, nasopharyngeal, or oropharyngeal obstruction and can be heard with a variety of congenital conditions described below. Nasal causes of obstruction can be particularly severe since most infants are obligate nasal breathers for the first several weeks to months of life. Stridor is higher pitched than sterdor and arises from turbulent airflow passing through an obstructed larynx or trachea. Correlating the stridor’s relationship to the respiratory cycle helps to identify the site of obstruction: inspiratory stridor arises from constricted airflow above the vocal cords; expiratory stridor arises from obstruction below the vocal cords; and biphasic stridor usually arises from lesions at the level of the vocal cords. Interestingly, the most commonly recognized causes of stridor in infants including laryngomalacia, subglottic stenosis, and subglottic hemangiomas, do not manifest immediately upon birth but appear days to months later. Stridor present immediately upon delivery is nearly always due to one of the three causes: an obstructing laryngeal cyst, a glottic web, or bilateral vocal cord paralysis; all conditions that, in severe cases, may require emergent airway control with intubation, laryngeal mask, or tracheotomy prior to investigating the actual cause of the obstruction.
Neonates with stridor who do not require emergent airway management should undergo a bedside flexible fiberoptic nasopharyngolaryngeal examination to assess the airway to the level of the vocal cords. Cardiac and pulse oximetry monitoring are advisable and blow by oxygen should be available, although infrequently needed. Lesions below the vocal cords arising from the subglottis, trachea, or bronchi require rigid bronchoscopy under general anesthesia with spontaneous respirations so that airway dynamic can be assessed. Direct visualization under general anesthesia is superior to imaging by MR, CT, cervical radiographs, or ultrasound. The infant’s unique ability to simultaneously breathe and suckle is often hindered by airway obstruction and some degree of feeding difficulty typically accompanies airway anomalies. A modified barium swallow study or bedside Feeding Endoscopic Evaluation of Swallowing (FEES) with a speech pathologist is indicated, if concerns of aspiration are present.
If respiratory distress from the obstruction is severe, emergent airway intervention is necessary even before establishing the cause of the airway condition. Respiratory distress may be managed with
blow by oxygen, mask ventilation, placement of a laryngeal mask airway (LMA), or endotracheal intubation. An LMA is preferable to intubation for neonates in whom direct visualization of the larynx is difficult, notably those with micrognathia, midface hypoplasia, macroglossia, or cervical spine anomalies. A neonate may be intubated over a small fiberoptic flexible telescope passed through the LMA, if its oral side has been modified to allow passage of an endotracheal tube. The mean diameter of the subglottis of a term neonate of normal weight is 4.5 mm so should easily accommodate a 3.0 oral endotracheal tube, which has an outer diameter of 4.3 mm (7). Preterm babies should be intubated with a smaller sized tube. Uncuffed tubes are much preferred over cuffed tubes and a peritubal leak should be present at pressures of 25 to 30 mm Hg to reduce the risk of acquired subglottic or tracheal stenosis.
blow by oxygen, mask ventilation, placement of a laryngeal mask airway (LMA), or endotracheal intubation. An LMA is preferable to intubation for neonates in whom direct visualization of the larynx is difficult, notably those with micrognathia, midface hypoplasia, macroglossia, or cervical spine anomalies. A neonate may be intubated over a small fiberoptic flexible telescope passed through the LMA, if its oral side has been modified to allow passage of an endotracheal tube. The mean diameter of the subglottis of a term neonate of normal weight is 4.5 mm so should easily accommodate a 3.0 oral endotracheal tube, which has an outer diameter of 4.3 mm (7). Preterm babies should be intubated with a smaller sized tube. Uncuffed tubes are much preferred over cuffed tubes and a peritubal leak should be present at pressures of 25 to 30 mm Hg to reduce the risk of acquired subglottic or tracheal stenosis.
Nasal Obstruction
Nasolacrimal Duct Cysts
Nasolacrimal duct cysts are the intranasal manifestation of a dacrocystocele and arise from the inferior meatus under the inferior turbinate. Bilateral cysts or unilateral cysts with contralateral cyclic mucosal congestion can lead to respiratory distress in the newborn infant. Surgical excision or marsupialization of the cyst is indicated and the urgency is dependent on the degree of respiratory compromise.
Pyriform Aperture Stenosis
Pyriform aperture stenosis (PAS) is a rare entity in which overgrowth of the medial nasal process of the maxilla leads to nasal obstruction (Fig. 41.4). The pyriform aperture is the narrowest region of the nasal airway, and thus, small decreases in the crosssectional area can significantly decrease nasal airflow. Symptoms may range from mild nasal congestion, to difficulty feeding, to periods of apnea and respiratory distress. If conservative measures of nasal humidification with saline and decongestion with oxymetazoline fail, surgical excision of the bony overgrowth is indicated, and is often combined with postoperative nasal stenting (8). PAS can be associated with a central maxillary incisor, which may be present with other congenital midline anomalies.
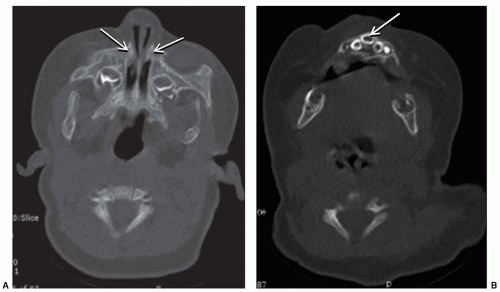 FIGURE 41.4 A: CT-axial image of a patient with pyriform aperture stenosis (PAS) (arrows). B: The same patient is also shown to have a central maxillary incisor (arrow). Patients with PAS should be evaluated for other midline abnormalities. |
Deviated Nasal Septum
A deviated nasal septum from intrauterine pressure or birth trauma may cause marked nasal obstruction in the newborn. The columella is tilted and anterior rhinoscopy demonstrates the septum deviated off the midline groove of the maxillary crest (9). External deformity of the nose may also be present. Radiographic imaging is not necessary as clinical exam is diagnostic. If acutely deformed during the delivery process but not displaced off the crest, the septum may return to the midline over the first several days of life. However, should the septum be deviated off the crest, gentle reduction under general anesthesia is indicated to improve breathing and to prevent the need for a septoplasty later in life. Nasal trauma may also cause a septal hematoma evident by a wide septum filling the nasal cavity. A septal hematoma requires emergent drainage to prevent cartilage destruction and resultant saddle nose deformity.
Choanal Atresia
Choanal atresia is the congenital absence of a communication between the posterior nasal cavity and the nasopharynx, most commonly caused by a bony plate at the posterior nasal choana. The incidence is approximately 1/5,000 to 8,000 live births. Diagnosis is suspected by inability to pass a 6-French catheter beyond 4 cm into the nasopharynx and is confirmed by CT scan (Fig. 41.5). It is critical to suction the nose immediately before the CT scan to differentiate mucus from an atretic plate. Females are affected more often, and unilateral disease, particularly on the right, is more common than bilateral disease. Importantly, 50% of patients with choanal atresia, particularly bilateral disease, will have other associated anomalies, the most common of which is
CHARGE association (coloboma, heart defects, choanal atresia, growth and mental retardation, genitourinary anomalies, and ear anomalies). Unilateral atresia usually presents with recalcitrant purulent rhinorrhea in infants or toddlers rather than neonatal airway obstruction. Unlike unilateral disease, bilateral atresia causes acute and severe respiratory distress in the neonate. Surgery is indicated to open the posterior choana(e) and establish a connection to the nasopharynx; and in bilateral cases, this is often urgent. Endoscopic techniques are most commonly used and postoperative stenting is controversial (10). A McGovern nipple forces the neonate to breathe through the mouth until surgery can be completed and is a viable alternative to intubation.
CHARGE association (coloboma, heart defects, choanal atresia, growth and mental retardation, genitourinary anomalies, and ear anomalies). Unilateral atresia usually presents with recalcitrant purulent rhinorrhea in infants or toddlers rather than neonatal airway obstruction. Unlike unilateral disease, bilateral atresia causes acute and severe respiratory distress in the neonate. Surgery is indicated to open the posterior choana(e) and establish a connection to the nasopharynx; and in bilateral cases, this is often urgent. Endoscopic techniques are most commonly used and postoperative stenting is controversial (10). A McGovern nipple forces the neonate to breathe through the mouth until surgery can be completed and is a viable alternative to intubation.
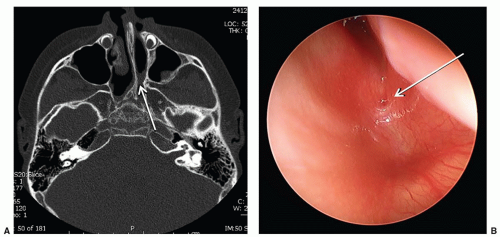 FIGURE 41.5 A: CT-axial image of a patient with left choanal atresia (arrow). Nasal cavity should be suctioned by radiology immediately prior to completing the scan. B: Correlative endoscopic view shows the blind ending at the choana (arrow). |
Congenital Midline Nasal Masses
Midline nasal masses include dermoids, encephaloceles, and gliomas that arise due to faulty embryogenesis in the 3rd week of gestation and may appear as a bulge in the midline nose, lateral nasal bridge, or forehead or as a pit anywhere along the nasal dorsum. Intracranial communication can occur with any of these anomalies. Transillumination, compressibility, and enlargement of the mass with crying are suggestive of an encephalocele with cerebrospinal fluid (CSF) communication. Gliomas and dermoids are solid lesions so lack communication with the CSF but may still have an intracranial stalk in up to 45% of cases. Surgical excision of midline nasal masses is indicated and requires careful preoperative planning and collaboration with neurosurgery to determine if an intracranial communication is present. A child with a midline nasal lesion should undergo both MR and CT imaging since the studies complement each other. The finding of a large foramen cecum, wide septum, and bifid crista galli on CT scan imply an intracranial connection even if one is not evident on MR imaging. Early surgical excision is indicated for encephaloceles due to the risk of meningitis otherwise excision or other midline masses may be deferred until preschool age.
Nasal Obstruction without Choanal Atresia
Sometimes referred to as “NOWCA,” this obstruction is felt secondary to nasal mucosal swelling and is usually a self-limited condition treated with saline drops, topical decadron, and tincture of time. It can be very symptomatic for the obligate nasal breather but does not require surgical intervention.
Oropharyngeal Obstruction
Congenital Tongue Cysts/Lesions
Neonatal tongue lesions can cause varying degrees of difficulty feeding and respiratory distress. While numerous types of lesions can present on the tongue, this section focuses on congenital cysts/lesions that can cause respiratory compromise.
Foregut duplication cysts are more common in males lined by a foregut-derived epithelium, and are most frequently seen in the oral cavity, although they can appear anywhere along the alimentary tract (11).
Vallecular cysts arise just anterior to the epiglottis and can displace the tongue base posteriorly leading to neonatal stridor or stertor (Fig. 41.6). These squamous epithelium-lined cysts are
thought to be formed from ductal obstruction of mucus glands and should be excised or marsupialized shortly after diagnosis as they can enlarge rapidly during the newborn period (12).
thought to be formed from ductal obstruction of mucus glands and should be excised or marsupialized shortly after diagnosis as they can enlarge rapidly during the newborn period (12).
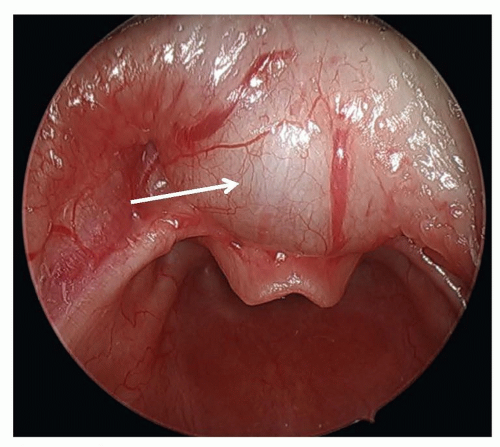 FIGURE 41.6 Preoperative endoscopic view of a vallecular cyst (white arrow). Note its position as well as its association with the tongue base and displacement of the epiglottis. |
Congenital epulis or congenital granular cell tumor is a benign soft tissue tumor most often affecting the alveolar mucosa of females but sometimes arising from the tongue where large masses can cause feeding difficulties or respiratory disturbance (13). While many tumors may regress during the first several months of life, surgical excision is indicated for symptomatic lesions, or if the diagnosis is unclear.
Thyroglossal duct cysts and lymphatic malformations can present as cystic lesions of the tongue, and management is discussed in the cervical section of this chapter.
Glossoptosis
Pierre-Robin sequence (PRS) consists of micrognathia, glossoptosis, and U-shaped cleft palate; it represents the best-known cause of glossoptosis or downward displacement of the tongue. Once considered a syndrome, PRS is now known to represent a sequence of events that begins with arrest in mandible development between the 7th and 11th week of gestation. The resultant micrognathia causes the tongue to rest higher in the oral cavity and prevent fusion of the palatal shelves (14). Genetic consultation is advised to exclude a syndrome, the most common of which is Stickler’s.
Airway obstruction in PRS is more severe in the supine position and worsens during sleep or induction of anesthesia. Airway obstruction can be managed by prone positioning, placement of a nasopharyngeal airway, intubation, tongue lip adhesion, or tracheotomy placement depending on the severity of the obstruction (15). Infants undergoing intubation should not be sedated until the airway is secure. Most patients without other significant airway or neurologic deficits can be managed conservatively without surgical intervention (16). As a general guideline, airway obstruction in PRS generally improves significantly by the time the baby doubles his/her birth weight. Infants with severe obstruction requiring a tracheotomy tube usually maintain the tube until the cleft palate repair is completed, thereby ensuring a secure airway for the surgical repair since intubation in these infants can be very difficult.
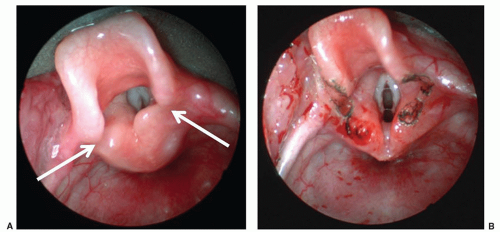 FIGURE 41.7 A: Classic endoscopic view of a patient with severe laryngomalacia. Note the shortened aryepiglottic folds (white arrows) as well as the supraglottic prolapse limiting visualization of the underlying vocal cords. B: Laser supraglottoplasty has been performed relieving the cause of obstruction and creating better visualization of the vocal cords. |
Macroglossia
Macroglossia may occur as an isolated anomaly or as a component of Beckwith-Wiedemann syndrome (BWS). This prenatal overgrowth syndrome most commonly arises from mutations of genes within the 11p15.5 region (17), and its hallmark findings include increased growth, abdominal wall defects, visceromegaly (particularly adrenal glands), hypoglycemia, and embryonal malignancies including Wilms tumor and hepatoblastoma (18). Tongue reduction via surgical excision or coblation can be considered; however, in symptomatic cases, a tracheotomy is often preferred since the obstruction improves when growth velocity normalizes later in the first decade of life.
True macroglossia and relative macroglossia (a small oral cavity) can also be seen in neonates with Down syndrome; however, the size of the tongue rarely necessitates surgical intervention.
Laryngeal Anomalies
Laryngomalacia
Laryngomalacia is the most common cause of stridor in infants and usually presents after several days of life. Inspiratory collapse of supraglottic structures, particularly the mucosa overlying the arytenoid cartilages and the aryepiglottic folds, leads to varying degrees of airway obstruction (Fig. 41.7). Infants may also have concomitant feeding difficulties and/or aspiration. Symptoms worsen with agitation and improve with prone positioning. Diagnosis is readily made by flexible fiberoptic laryngoscopy. Most infants can be managed conservatively as symptoms generally resolve by 12 to 24 months of age. Reflux medications are often utilized as some believe that reflux causes edema of the mucosal structures that leads to redundancy; however, the authors find them to be effective in only relatively small percent of children. Fifteen percent of babies with laryngomalacia require surgical intervention, more frequently due to failure to thrive than respiratory distress (19). Supraglottoplasty, in appropriately selected patients, is highly successful with very few complications (20). Tracheotomy is sometimes necessary, particularly in infants with multiple comorbid or underlying neurologic conditions that reduce the success rate of supraglottoplasty.
Vocal Cord Paralysis
Vocal cord paralysis is the second most common cause of stridor in infants following laryngomalacia. Stridor and other symptoms vary considerably based on whether the lesion is unilateral or bilateral.
Unilateral Vocal Cord Paralysis
Patients with unilateral vocal cord paralysis typically present with stridor (75%), dysphonia (weak or hoarse cry) (50%), and/or feeding difficulties (25%) (21). Early surgical intervention is rarely required. The most common etiology of unilateral paralysis is iatrogenic, following cardiothoracic surgery or tracheoesophageal fistula (TEF) repair and with the left vocal cord more commonly affected than the right. Spontaneous recovery occurs in a third of these patients but may not be evident until late in the first decade of life. Many cases are idiopathic, but other causes of unilateral paralysis include trauma (i.e., complicated delivery), intubation injury, neurologic conditions, and infection. Compensation by the contralateral mobile cord usually obviates the need for medialization procedures, but surgical treatment can be performed later in life if cord recovery does not occur and hoarseness is severe.
Bilateral Vocal Cord Paralysis
Prominent stridor and a loud cry are the hallmark features of bilateral vocal cord paralysis since the vocal cords are situated in the paramedian position. All patients with bilateral cord paralysis should undergo MR imaging to exclude Chiari malformation or other causes of brainstem compression. Other etiologies include neurologic, iatrogenic, traumatic, and idiopathic causes the latter carrying the greatest prognosis for spontaneous recovery. Bilateral is less common than unilateral paralysis but more often requires airway management with approximately 50% requiring tracheotomy (22). Arytenoidectomy can enhance success of decannulation, but is usually deferred for several years of life to allow potential for spontaneous recovery.
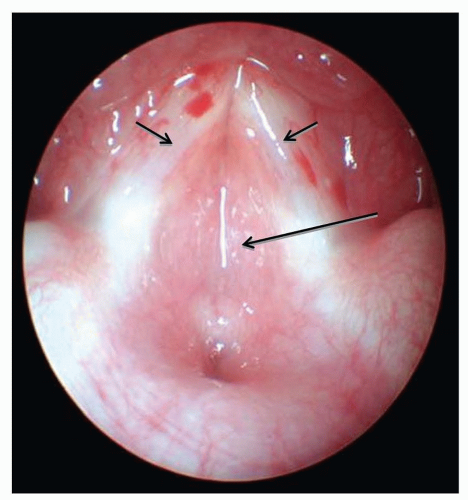 FIGURE 41.8 Severe congenital laryngeal web. This patient required emergent tracheotomy tube placement. Prenatal screening would have demonstrated CHAOS, at which time an EXIT procedure could have been planned (small arrows indicate vocal cords, large arrow indicates glottis completely obstructed by congenital web). |
Laryngeal Atresia or Web
Complete laryngeal atresia is a very rare congenital anomaly, which results from failure of the larynx and trachea to recanalize during the 7th to 8th week of gestation (23) (Fig. 41.8). Patients present with severe respiratory distress despite adequate effort. Prenatal ultrasound may identify the developmental sequelae of CHAOS (congenital high airway obstruction syndrome), which includes polyhydramnios, dilated airway(s) distal to obstruction, enlarged lungs, fetal hydrops, and/or a flattened diaphragm (24). If this syndrome is diagnosed prenatally, an ex utero intrapartum treatment (EXIT) procedure should be coordinated to secure the airway. If not diagnosed prenatally, an emergent tracheotomy is necessary.
Congenital laryngeal webs are a form of laryngeal atresia where partial canalization has occurred. The failure to fully recanalize the larynx results in various degrees of webbing most commonly at the level of the vocal cords with extension to the subglottis. These patients can present along a spectrum of severity from an isolated weak cry to varying degrees of respiratory compromise. Anterior glottic webs can occur in conjunction with other congenital anomalies, particularly cardiovascular and chromosomal defects. Only webs causing significant symptoms need to be addressed emergently. Otherwise, management is usually deferred until after 1 year of age and entails dilation or endoscopic micro- or laser division or an open laryngoplasty (25).
Laryngeal Cleft
Laryngeal clefts are rare anomalies that arise when there is a lack of separation between the laryngotracheal and pharyngesophageal systems due to inadequate development of the tracheoesophageal septum. Consequently, a posterior defect in the laryngotracheal apparatus communicates directly with the hypopharynx and/or esophagus (Fig. 41.9). Laryngeal clefts are best described as type I, II, III, or IV, with the latter types being more severe (26). Type I clefts may be managed medically in some patients, but types II to IV will require surgical intervention, often through an open approach. These patients present with a spectrum of respiratory and feeding difficulties commensurate with the severity of their cleft. Aspiration, chronic cough, stridor, respiratory distress, and recurrent pneumonia are all potential presenting symptoms. Laryngeal clefts are associated with other congenital anomalies in close to 60% of cases making thorough evaluation and workup essential.
Subglottic Stenosis and Intubation Trauma
Subglottic stenosis is the third most common cause of stridor in the pediatric population following laryngomalacia and vocal cord paralysis (Fig. 41.10). A subglottic diameter of less than 4 mm in a term infant and less than 3 mm in a preterm infant is considered to be narrowed. This airway lesion is best classified into congenital and acquired types.
Congenital Subglottic Stenosis
Subglottic stenosis is considered congenital when there is no history of endotracheal intubation or other potential cause of the subglottic narrowing. The stenosis can be a result of fibrous soft tissue thickening or can be from a cartilaginous deformity of the cricoid plate. Patients may present with inspiratory stridor that can escalate to biphasic stridor in cases of higher-grade stenosis. Patients are prone to recurrent croup. Congenital subglottic stenosis is often less severe than the acquired type. While some patients can be managed conservatively and allowed to outgrow the lesion, others with higher-grade stenoses or with uncontrolled symptoms will require surgical correction with a laryngotracheoplasty or tracheostomy.
Acquired Subglottic Stenosis
Trauma from prolonged or traumatic intubation is the most common cause of acquired subglottic stenosis. The incidence of this pathology in intubated neonatal intensive care babies is less than 2% (27). The size of the endotracheal tube and the duration of intubation are important factors in the development
of acquired subglottic stenosis. While there is no clear evidence to suggest that rates of acquired subglottic stenosis are different between orotracheal and nasotracheal intubation, it is quite clear that nasotracheal intubation is better tolerated by infants. The need for surgical intervention is highly dependent on the patient and the degree of stenosis. Balloon dilation or airway reconstruction may be attempted in select patients; however, if the patient has significant comorbidities (i.e., chronic lung disease) and is unable to be extubated, a tracheostomy may be indicated with plans to address the subglottic stenosis once the baby has grown.
of acquired subglottic stenosis. While there is no clear evidence to suggest that rates of acquired subglottic stenosis are different between orotracheal and nasotracheal intubation, it is quite clear that nasotracheal intubation is better tolerated by infants. The need for surgical intervention is highly dependent on the patient and the degree of stenosis. Balloon dilation or airway reconstruction may be attempted in select patients; however, if the patient has significant comorbidities (i.e., chronic lung disease) and is unable to be extubated, a tracheostomy may be indicated with plans to address the subglottic stenosis once the baby has grown.
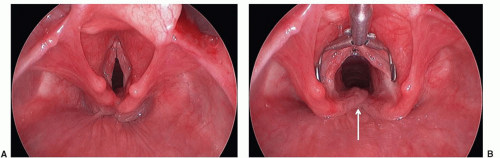 FIGURE 41.9 A: Endoscopic view of a patient with a laryngeal cleft. The lesion is difficult to fully appreciate prior to spreading the vocal cords. B: With the vocal cords distracted, the posterior cleft can be appreciated extending below the level of the vocal cords (white arrow). |
Subglottic Hemangioma
Infantile hemangiomas are the most common tumor of infancy and can occasionally arise in the subglottis. Infants with subglottic hemangiomas are typically asymptomatic until the hemangiomas enter the proliferative phase, usually after 4 weeks of age. The resultant stridor can be inspiratory or biphasic and, like stridor from other causes, is more pronounced with feeding and exertion. Uniquely, stridor arising from subglottic hemangiomas worsens in the supine position as the lesion engorges with blood and becomes more obstructive. In rare cases, stridor is absent and wheezing, barky cough or hoarseness may be the presenting symptoms. Cutaneous hemangiomas are present in 50% of cases. The incidence of subglottic hemangiomas is much higher in patients with a beard-like distribution of hemangiomas, particularly if bilateral. Diagnosis is best made by prompt direct laryngoscopy (Fig. 41.11), although plain neck films will show a smooth subglottic asymmetric narrowing and can be helpful. MR or CT is indicated to determine if mediastinal extent is present. Like cutaneous hemangiomas, subglottic hemangiomas eventually involute, but unlike cutaneous hemangiomas, watchful waiting or observation is not acceptable since the airway obstruction from these lesions can be fatal. The degree of airway narrowing in a relatively asymptomatic infant with a subglottic hemangioma is always remarkable.
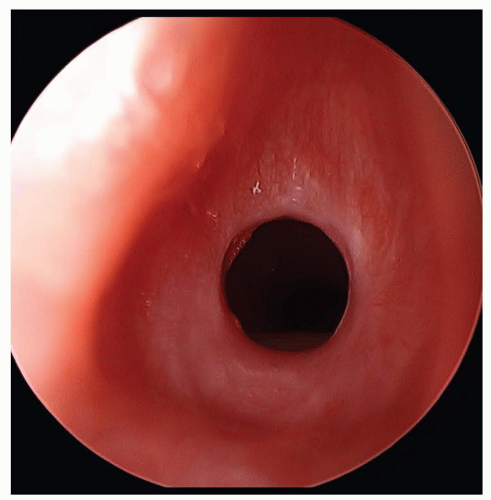 FIGURE 41.10 Acquired subglottic stenosis from traumatic intubation. Note the mature circumferential scar. |
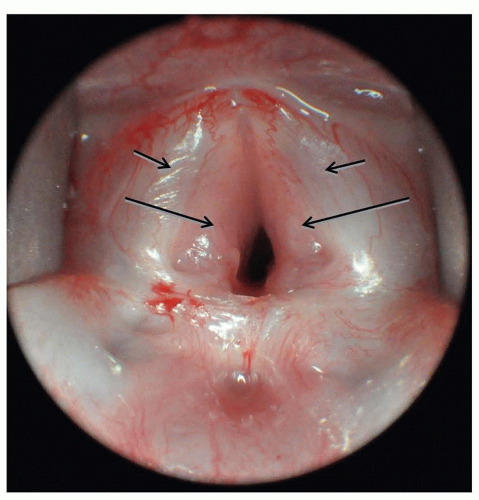 FIGURE 41.11 Large bilateral subglottic hemangioma (large arrows) immediately adjacent to vocal cords (small arrows) causing progressively worsening stridor. |
Propranolol is effective monodrug treatment for most subglottic hemangiomas (28) and should be initiated as an inpatient in these young infants with airway lesions (29). For symptomatic infants, intravenous corticosteroids are often given concomitantly for the first 2 to 3 days of treatment. Oral corticosteroids are an alternative monodrug treatment to propranolol, but are no longer considered first line of therapy due to the high rate of predictable adverse events, particularly temporary growth restriction (30). Similarly, IFN-2a is no longer recommended for hemangioma treatment due to the risk of spastic diplegia when administered to young infants (31). For markedly obstructive lesions, intubation during the first week of pharmacologic therapy may be necessary. Repeat laryngoscopy to assess the hemangioma’s response to therapy is advisable for large lesions. Surgical excision or endoscopic laser resection is effective alternative for unilateral lesions that do not respond to pharmacologic therapy, or in whom pharmacologic therapy may be contraindicated. Tracheotomy is sometimes necessary for large, bilateral lesions, but a 2-week trial of intubation and pharmacologic therapy first should be considered to try to avoid tracheotomy tube placement with its attendant social and medical issues.
Tracheal Obstruction
Tracheal Stenosis
Tracheal stenosis may be acquired secondary to endotracheal tube intubation, particularly if a cuffed tube is used, but most cases are congenital in origin. Congenital tracheal stenosis may involve only a short segment of the trachea or the entire trachea and can extend into the bronchus as well. Most segments of stenosis are complete tracheal rings that lack a posterior membranous wall (Fig. 41.12) and in nearly 50% of cases are associated with a pulmonary sling (32). Symptoms vary widely from minimal to life-threatening respiratory distress and when stridor is present, it is often expiratory, low-pitched, and often mimics the sound of the agitation cycle of a washing machine. Cough, wheezing, and feeding difficulties are also common presenting symptoms all of which may worsen following an upper or lower respiratory tract infection. Endoscopy is the preferred method of diagnosis, although chest radiographs, MR, and CT can all demonstrate the narrowing. Endotracheal intubation is avoided when possible as even a small amount of edema in the narrowed lumen may prevent extubation. Mortality of 12% is noted either from associated heart, vascular, or lung anomalies or from complications of surgical repair (32). Mild cases can be managed expectantly with steroids, bronchodilators, and physiotherapy, but more severe cases require surgical intervention entailing resection with end-to-end anastomosis, costal cartilage tracheoplasty, balloon dilation, or, most commonly, slide tracheoplasty (33). Multiple trips to the operating room should be anticipated to treat postoperative granulation tissue.
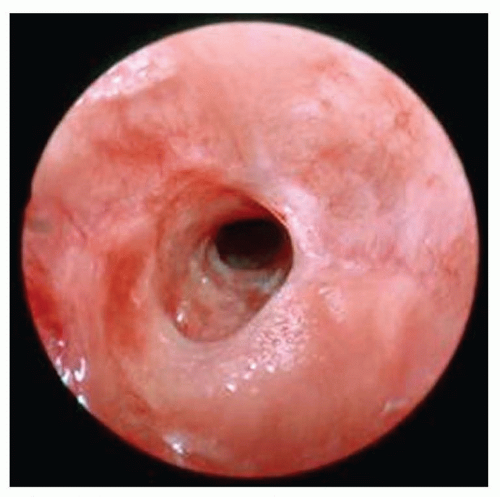 FIGURE 41.12 Acquired tracheal stenosis from traumatic intubation. Note the multilevel stenosis with circumferential scarring. |
Tracheomalacia
The 4.5:1 cartilaginous:membranous ratio of the normal horseshoe-shaped trachea is decreased in infants with tracheomalacia and the wide posterior membranous wall allows collapse of the trachea (Fig. 41.13). Tracheomalacia may be primary (intrinsic)
or secondary (extrinsic) due to external compression. Virtually, all patients with TEF have some degree of tracheomalacia. Expiration is compromised and symptoms vary widely in their severity and include chronic cough, wheezing, recurrent pneumonia, respiratory distress, and, in some cases, reflex apnea. The resultant cough is “barky” in nature due to the turbulent airflow created by the collapsing trachea. Tracheomalacia is evident on cine CT in a nonintubated infant, but the diagnosis is best made by bronchoscopy under spontaneous ventilation. Medical management includes chest physiotherapy and antibiotics for exacerbations as the trapped secretions often become secondarily infected. CPAP or bipap is useful in maintaining tracheal patency. Although infants outgrow tracheomalacia, often by 2 years of age, surgical intervention with either tracheotomy or aortopexy is indicated for severe cases that demonstrate reflex apnea, recurrent respiratory distress, or recurrent pneumonia.
or secondary (extrinsic) due to external compression. Virtually, all patients with TEF have some degree of tracheomalacia. Expiration is compromised and symptoms vary widely in their severity and include chronic cough, wheezing, recurrent pneumonia, respiratory distress, and, in some cases, reflex apnea. The resultant cough is “barky” in nature due to the turbulent airflow created by the collapsing trachea. Tracheomalacia is evident on cine CT in a nonintubated infant, but the diagnosis is best made by bronchoscopy under spontaneous ventilation. Medical management includes chest physiotherapy and antibiotics for exacerbations as the trapped secretions often become secondarily infected. CPAP or bipap is useful in maintaining tracheal patency. Although infants outgrow tracheomalacia, often by 2 years of age, surgical intervention with either tracheotomy or aortopexy is indicated for severe cases that demonstrate reflex apnea, recurrent respiratory distress, or recurrent pneumonia.
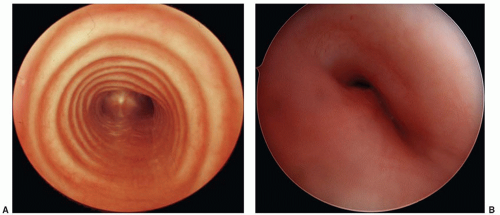 FIGURE 41.13 A: Normal trachea exhibiting well-defined cartilaginous rings and membranous posterior wall. B: Tracheomalacia with collapse of the airway distally secondary to vascular compression from a double aortic arch. Note large membranous wall component. |
Vascular Compression of the Trachea
Abnormal development of the aortic arch and its main vessels typically causes tracheal obstruction and infants present with chronic cough, dyspnea, expiratory stridor, wheezing, reflex apnea, and, in some cases, dysphagia. Anomalies are best visualized by bronchoscopy often combined with esophagoscopy. Magnetic resonance imaging with angiography (MRA) or computed tomography with angiography (CTA) are reserved for cases requiring surgical intervention.
The anomalous innominate artery, the most common symptomatic anomaly (34), originates more distally from the arch of the aorta and compresses the trachea as it crosses from left to right. Surgical therapy with aortopexy is indicated for severe cases with recalcitrant symptoms.
▪ THORACIC LESIONS CAUSING RESPIRATORY DISTRESS
Congenital Lobar Emphysema
Congenital lobar emphysema (CLE) usually affects one lobe and can be life threatening as a result of severe hyperexpansion. The underlying cause of the disorder is air entry into the affected lobe on inspiration and air-trapping during expiration. Lobar overexpansion occurs with compression of adjacent pulmonary parenchyma and mediastinal displacement. CLE can occur as the result of bronchial stenosis, bronchiomalacia, luminal obstruction, or extrinsic compression of the airway by a mass or bronchogenic cyst. A chest radiograph shows hyperlucency of the affected lobe associated with atelectasis of the adjacent normal lung and mediastinal shift away from the affected side (Fig. 41.14).
The left upper lobe is the most commonly affected lobe followed by the right middle lobe and then the right upper lobe. The lesion is rarely bilateral (35). The management of CLE depends on the presence of symptoms. If asymptomatic or only mildly symptomatic, patients can be successfully observed without requiring surgical resection. For patients with progressive respiratory distress, emergent thoracotomy and lobectomy are required. Patients with CLE can also develop persistent or recurrent pneumonias requiring elective resection.
Congenital Pulmonary Airway Malformation
Congenital pulmonary airway malformation (CPAM) refers to a group of lesions characterized by the abnormal development of pulmonary parenchyma. Lesions with cystic replacement of lung parenchyma are also known as congenital cystic adenomatoid malformations (CCAMs) in older terminology. CCAMs usually arise from a single lobe of the lung and most commonly involve the left lower lobe. Lesions with a systemic arterial feeding vessel are called bronchopulmonary sequestrations (BPS). This nonfunctional pulmonary tissue can be intralobar, extralobar, or intra-abdominal. Lesions with both cystic features and systemic arterial blood supply are termed hybrid lesions.
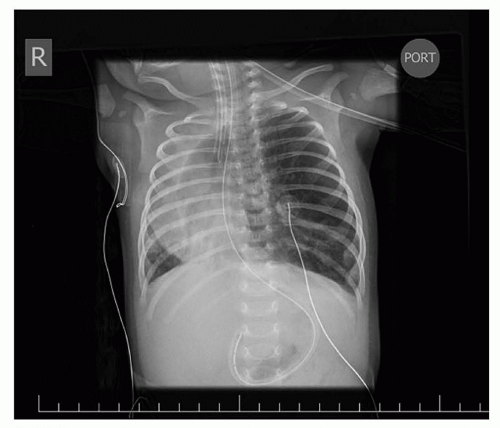 FIGURE 41.14 A chest radiograph shows hyperlucency of the affected lobe associated with atelectasis of the adjacent normal lung and mediastinal shift away from the affected side. |
CPAMs are most commonly diagnosed by prenatal ultrasound. The cyst volume ratio (CVR) was developed to help risk stratify lesions. The CVR is the ratio of the volume of the congenital airway malformation divided by the head circumference as measured by prenatal ultrasound. Lesions with a CVR greater than 1.6 are associated with a higher risk for fetal hydrops (36). Since CPAMs have the capacity for unpredictable growth during midgestation, fetuses must be followed closely with serial ultrasound to document interval growth of the lesion until it plateaus and the normal lung growth exceeds that of the lesion. Fetuses with high-risk lesions are candidates for in utero maternal steroid administration to arrest growth of the lung lesion (37). High-risk lesions that do not respond to maternal steroids and are associated with hydropic changes may be candidates for fetal resection of the lesion. Large macrocystic lesions causing hydrops are best treated with in utero thoracoamniotic shunt placement (38).
Most pediatric surgeons recommend postnatal surgical resection of these lesions because of the risk of infection and malignancy (39). The timing of resection is based on the presence of symptoms at birth. Most infants are born asymptomatic and resection can be preformed electively between 2 and 6 months of age via a thoracoscopic approach or thoracotomy (40). CT scan of the chest with contrast is obtained postnatally to confirm the diagnosis and identify any systemic feeding vessels prior to surgery (Fig. 41.15). Infants with symptomatic lesions require immediate resection of the involved lobe; and this is typically preformed via thoracotomy because of the larger size of these lesions.
Congenital Diaphragmatic Hernia
Failure of the development of the posterolateral portion of the diaphragm results in persistence of the pleuroperitoneal canal or foramen of Bochdalek, which allows the abdominal viscera to occupy space in the chest cavity creating a mass effect that impairs normal lung development. Eighty percent of defects occur on the left side and 20% occur as right-sided defects. The pathophysiology of Congenital diaphragmatic hernia (CDH) relates to the resultant pulmonary hypoplasia. The hypoplastic lung is not only small but lacks the normal bronchiole branching pattern, alveolar surface area,
and pulmonary vascular structure (41). The pulmonary arteries are hypermuscular with increased pulmonary vascular resistance and reactivity, characteristic of pulmonary hypertension.
and pulmonary vascular structure (41). The pulmonary arteries are hypermuscular with increased pulmonary vascular resistance and reactivity, characteristic of pulmonary hypertension.
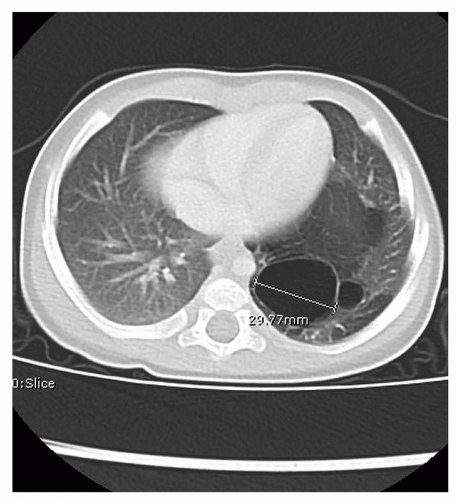 FIGURE 41.15 CT scan of the chest at 6 months of age showing cystic left lower lobe CPAM. |
The majority CDH cases are diagnosed by prenatal ultrasound. Sonographic findings include herniated abdominal viscera, mediastinal shift with abnormal heart position, or pleural effusion (42). Right-sided defects are harder to diagnose prenatally, because it is difficult to distinguish herniated liver from the right lung. Various prenatal lung measures have been developed to help stratify patients and predict postnatal outcomes (e.g., lung-head ratio [LHR], observed to expected LHR, and observed to expected total lung volumes) (43). Prenatal diagnosis allows for optimal delivery planning. Delivery of infants with CDH should take place at institutions with experience in support of these infants and access to pediatric surgical support and extracorporeal membrane oxygenation (ECMO). The overall goal in delivery room management and transport is to minimize perinatal stress and prevent events that trigger pulmonary vasospasm and the clinical decline that is difficult to reverse.
Infants with known CDH are immediately intubated at birth and a nasogastric tube placed to decompress the bowel. Infants with CDH who have not been diagnosed prenatally present with respiratory distress at birth and the diagnosis is made by the presence of bowel within the chest on x-ray (Fig. 41.16). Infants with small hernia defects may be asymptomatic at birth. In these cases, CDH is often diagnosed incidentally when a chest x-ray is obtained for another reason.
The initial goal in management is support of gas exchange and prevention of a pulmonary hypertensive crisis. Permissive hypercarbia and gentle ventilation have proven effective in maintaining adequate oxygenation and ventilation without injuring the hypoplastic lungs. Surgical repair of the diaphragm is delayed until the pulmonary hypertension resolves or at least until it improves significantly and the infant requires less mechanical ventilatory support.
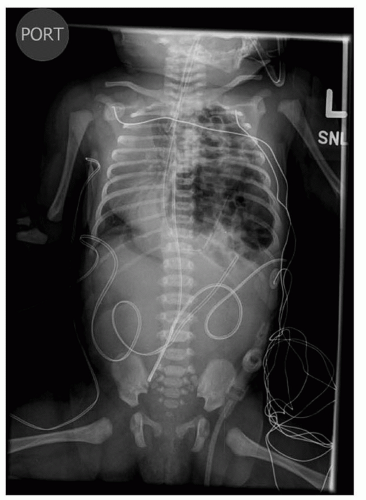 FIGURE 41.16 Chest x-ray demonstrating bowel herniating into the left chest with mediastinal shift. Nasogastric tube is seen in the left chest. |
ECMO is used to support the CDH infant only when standard therapy fails. Venoarterial bypass is used and continued until the pulmonary hypertension is reversed and lung function is improved. Most infants improve within 7 to 10 days, but some may require longer support. Failure to improve after 2 to 3 weeks is indicative of severe pulmonary hypoplasia and may not be survivable.
Surgical repair is generally preformed via an open abdominal approach. The surgeon reduces the abdominal contents from the chest back into the abdomen and the edges of the diaphragm are identified. If adequate diaphragmatic tissue is available, the edges are sutured together primarily. If diaphragm tissue for repair is lacking, prosthetic material is used to fashion a patch to reconstruct the diaphragm and close the defect. After CDH repair, ventilatory support is gradually weaned and enteral feedings initiated.
The reported survival for isolated CDH ranges from 70% to 90% overall with survival rates decreasing to 50% when patients require ECMO. Infants with severe CDH have a higher incidence of associated respiratory, neurologic, GI and nutritional morbidity (44,45,46,47). Many high volume centers have created specialized multidisciplinary follow-up clinics to address the complex needs of this population of survivors (48).
Eventration of the Diaphragm
Eventration of the diaphragm may be congenital or acquired. The congenital presentation may mimic that of a CDH with a sac. The acquired lesion results from paralysis of the diaphragm, most commonly caused by operative trauma or birth injury (49).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


