BIOPSY DIAGNOSIS OF SOFT TISSUE LESIONS
Biopsies of soft tissue tumors may be performed in several different ways. These include fine needle aspiration, large-needle core biopsy, open incisional biopsy, and excisional biopsy. The last technique, excisional biopsy, is generally reserved for superficial lesions that can be easily excised with adequate margins. In these lesions, it is critical to ink carefully the exterior surface of the lesions prior to fixation because marginal involvement upstages lesions and may lead to radiation therapy or additional surgery. In all lesions, some tissue should be set aside and frozen prior to fixation because patients with malignant tumors will likely be enrolled on a multi-institutional therapeutic trial or biology study that requires submission of fresh material. However, for small lesions or biopsies, it is more important to obtain sufficient material for diagnosis, and most of these trials will accept multiple paraffin sections if fresh tissue is unavailable (but as a less desirable substitute). For small-needle biopsies, the operator should then perform extra passes and save material in a small amount of tissue culture media such as RPMI. For deep-seated or large lesions, primary excision is rarely indicated because observation of tumors following chemotherapy is important to determine response, and pretreatment enhances the probability of complete tumor removal and surgical cure. Consideration should also be made of standard cytogenetic karyotyping, but this technique has been superseded in the majority of cases by techniques such as FISH or RT-PCR (which today is usually performed on paraffin sections), and the results are generally not available until after therapy has been initiated. If fresh tissue is frozen for biologic study and the material obtained for diagnosis is inadequate, then the biologic study materials can be formalin fixed and paraffin embedded for the all-important diagnosis, so it is wise to retain this material until the paraffin sections are judged to be sufficient.
At the time of frozen section diagnosis, it is always wise to prepare touch preparations of soft tissue lesions. Admittedly, touch preps can yield paucicellular or bare slides, but it is invaluable to correlate cytologic features with histology. In this manner, one can more easily separate eosinophils from rhabdomyoblasts, lymphocytes and plasma cells from “small blue round cell” tumors, and possible hematopoietic tumors from nonhematopoietic ones. For the latter separation, lymphomas and leukemias generally exhibit lymphoglandular bodies (fragments of apoptotic cells) and lack cohesion. If these features are present, flow cytometry and cytogenetics are in order.
Final diagnosis of soft tissue lesions should always be specific. Diagnoses such as “spindle cell tumor,” “round cell sarcoma,” and the like are no better for clinical management than “inadequate tissue for diagnosis” and should always be followed up with more studies, consultation, or additional biopsies. Chemotherapy, radiation therapy, and excisional surgery and prognostication are all based on knowledge of the behavior of specific entities. However, if a specific diagnosis is not forthcoming after an extensive workup, it is appropriate to use the terms “undifferentiated round cell sarcoma” or “undifferentiated pleomorphic sarcoma,” followed by tumor grade (see the WHO Classification of Tumours of Soft Tissue and Bone, soon to be revised to include undifferentiated lesions). Grading of pediatric sarcomas has mostly been performed with the Pediatric Oncology Group schema, but recent comparison with the French (or Fédération Nationale des Centres de Lutte Contre le Cancer [FNCLCC]) schema suggests that the latter has more predictive value. With the advent of advanced molecular studies, much more knowledge of undifferentiated sarcomas has been gained, and some have a characteristic DUX4-CIC translocation.
FIBROBLASTIC AND MYOFIBROBLASTIC TUMORS
Fibroblastic/myofibroblastic proliferations comprise a wide variety of spindle cell lesions, from reactive nonneoplastic lesions to benign, intermediate malignant, and malignant neoplastic tumors. Most lesions are benign with excellent outcome. Even apparent malignant tumors may have a better prognosis than their adult counterpart, for instance, infantile fibrosarcoma. Although their tumor-related deaths are low, some lesions tend to recur and require repeated surgeries, which may cause disfigurement or dysfunction of adjacent organs. Some tumors may be associated with congenital or familial cancer syndromes. This group of tumors requires appropriate treatment, follow-up, and genetic counseling.
Nodular Fasciitis
CLINICAL FEATURES. Nodular fasciitis (NF) often presents as a rapidly growing painless mass, raising clinical concerns for malignancy. Although it has been described virtually anywhere in the body, in children, NF particularly favors the head and neck region, most often in deep subcutaneous tissue, fascia, and muscle. Recurrence is rare even after incomplete excision.4
PATHOLOGIC FEATURES. NF forms a small (usually <3.5 cm), nonencapsulated soft tissue mass with an infiltrative border or adherence to adjacent structures. Histologically, plump, spindle-shaped fibroblasts/myofibroblasts are loosely arranged in a tissue culture–like pattern, with focal myxoid, torn, and feathery features or a vague storiform pattern mimicking fibrous histiocytoma. The spindle cells have ovoid to spindle-shaped nuclei without significant nuclear pleomorphism. Although there are usually numerous mitoses, none should be atypical. Extravasated red blood cells, scattered lymphocytes, and occasional multinucleated giant cells are often seen in the background (Fig. 4.1). Due to its hypercellularity, brisk mitoses, and immature appearance, NF is easily misdiagnosed as a pleomorphic-storiform sarcoma (malignant fibrous histiocytoma) especially on a limited biopsy, so one should be familiar with this entity and perform careful inspection.

Cranial fasciitis, a variant of NF, deserves special mention. It involves the scalp of young children, usually infants, and presents as a rapidly growing soft tissue mass eroding the skull and may involve the dura. In some cases, it may present intracranially.5
ANCILLARY STUDIES. IHC has limited use for diagnosis. Most lesions show positivity for smooth muscle actin (SMA), consistent with myofibroblastic differentiation. Some cells are also positive for CD68, a histiocytic marker.6 NF generally lacks nuclear staining for β-catenin except for cranial fasciitis, which may show diffuse nuclear positivity.7,8
Infantile Digital Fibromatosis
CLINICAL FEATURES. Infantile digital fibromatosis (IDF) typically presents in infants as a solitary, slow-growing reddish nodule on the lateral and dorsal aspects of the second to fifth fingers and toes. The tumor often recurs after incomplete surgery (74% recurrence rate), but it usually regresses with time.9
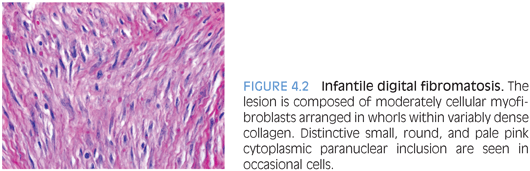
PATHOLOGIC FEATURES. IDF is typically small, forming a dome-shaped, firm, nonencapsulated dermal or subcutaneous nodule covered by intact skin. It consists of moderately cellular spindle-shaped myofibroblasts arranged in whorls or interlacing sheets within variably dense collagen (Fig. 4.2). Occasional cells contain a distinctive eosinophilic, cytoplasmic, paranuclear inclusion in occasional cells. The inclusions are usually slightly variable in size and nonrefractile, which distinguishes them from red blood cells. Large lesions may extend into the periosteum, but they seldom erode the bone.
ANCILLARY STUDIES. The inclusions stain red with the Masson trichrome stain (eFig. 4.1). The inclusions are typically actin-negative.10 The spindle cells are positive for actin, calponin, desmin, and several cytokeratins, variably positive for CD99, CD117, CD34, and CD10, and rarely (<5%) positive for nuclear β-catenin.9,11,12
Fibrous Hamartoma of Infancy
CLINICAL FEATURES. Fibrous hamartoma of infancy (FHI) typically occurs in the infants and toddlers and shows a predilection for males. Most patients present with a painless, firm subcutaneous nodule, sometimes with rapid growth. The commonest site is the axilla. Most tumors are solitary. The treatment of choice is local excision, with up to 16% recurrence.
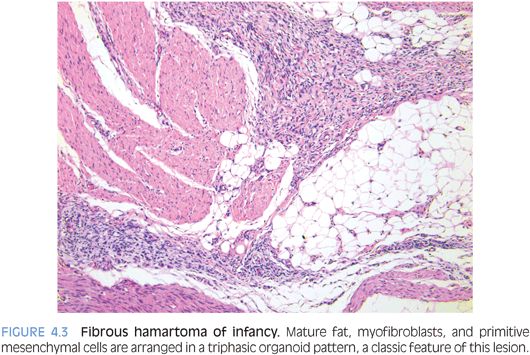
PATHOLOGIC FEATURES. FHI usually forms a poorly circumscribed, 1- to 8-cm mass in the subcutis. Histologically, FHI has a triphasic organoid pattern with intervening mature fat; streaks of moderately cellular fibroblastic areas; and small clusters of oval, more primitive mesenchymal cells (Fig. 4.3). Often, these primitive cells form concentric pattern with whorls or ball-like structures in a mucoid matrix. The relative proportions of the three components vary greatly.
ANCILLARY STUDIES. The spindle cells in FHI are positive for actin and sometimes desmin, suggesting myofibroblasts.
Lipofibromatosis
CLINICAL FEATURES. Lipofibromatosis (LF) is a hamartomatous tumor of infants and young children, with predilection to males. About 18% of cases occur at birth. Most patients present with a slow-growing mass, often in the hand or foot. The recurrence rate is high, up to 72%, if incompletely excised.13
PATHOLOGIC FEATURES. LF is a poorly circumscribed, 1- to 7-cm nonencapsulated fatty or fibrofatty subcutaneous mass. It contains two components, fat and fibrous tissue, that form a vague lobular architecture. The fibrous tissue comprises bundles of spindle-shaped fibroblasts and little to moderate collagen, with occasionally myxoid stroma. Tumor cells have no significant cytologic atypia, nuclear pleomorphism, or mitosis. The adipose tissue comprises mature fat with occasional univacuolated lipoblasts between fibroblastic areas and mature fat. Entrapped vessels, nerves, skin adnexa, and skeletal muscle are frequent. The overall histology resembles FHI except for the absence of primitive mesenchymal cell nests or an organoid pattern.
ANCILLARY STUDIES. The spindle cells of LF are often positive for CD99, CD34, and BCL-2, but they are negative for nuclear β-catenin.8,11,13
Desmoid Tumor (Desmoid-Type Fibromatosis, Aggressive Fibromatosis)
CLINICAL FEATURES. Desmoid tumor (DT) usually affects patients older than 5 years of age. There is no difference in tumor behavior between children and adults. Most are located in the head and neck, extremities, trunk, and hip regions. Most patients present with a slowly growing, nontender mass, which recurs frequently after incomplete excision.
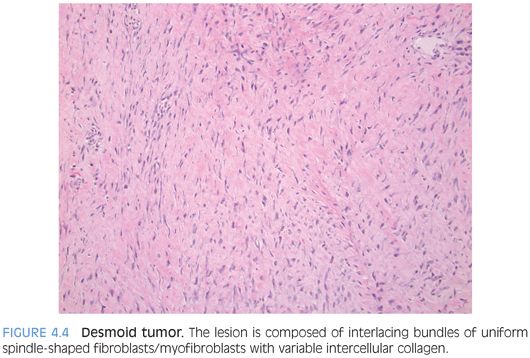
PATHOLOGIC FEATURES. DT varies from a small nodule to a bulky mass with a firm trabeculated appearance (eFig. 4.2). DT is composed of interlacing bundles of uniform spindle-shaped fibroblasts/myofibroblasts with variable intercellular collagen (Fig. 4.4). The cellularity is low to moderate with no significant cytologic atypia or mitosis. Slitlike vessels are often seen, sometimes accompanied with perivascular edema. DT often infiltrates into the surrounding skeletal muscle, tendon, and fat at the margin of resection.
ANCILLARY STUDIES. Virtually, all DTs have somatic β-catenin or germline adenomatous polyposis coli (APC) gene mutations, causing nuclear expression of β-catenin (eFig. 4.3).14 The spindle cells are also positive for SMA, consistent with myofibroblasts.
Gardner Fibroma
CLINICAL FEATURES. Gardner fibroma (GF) is a benign fibrous lesion that may serve as a sentinel lesion for patients with Gardner syndrome, familial adenomatous polyposis, and/or APC mutation. It tends to occur in young patients, mostly younger than 10 years of age. GF usually arises in the superficial and deep soft tissues, mainly the paraspinal region and back. About 50% have a coexisting or subsequent DT.15
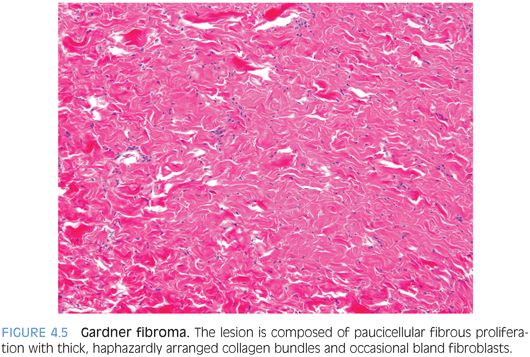
PATHOLOGIC FEATURES. GF forms a nonencapsulated, poorly demarcated, plaquelike soft tissue mass that histologically resembles nuchal-type fibromas and features a paucicellular fibrous proliferation with thick, haphazardly arranged collagen bundles and occasional bland fibroblasts (Fig. 4.5). It can look deceptively like normal or nonneoplastic tissue, but it forms a discrete mass.
ANCILLARY STUDIES. GF is positive for CD34 and negative for SMA and desmin. Nuclear β-catenin expression is variable; 64% in one study (16/25).15
Infantile Myofibroma
CLINICAL FEATURES. Infantile myofibroma (IM) is one of the most common fibrous tumors of infancy and childhood.16 Most patients have a solitary lesion that arises in the dermis, subcutis, or deep soft tissue, of the head and neck, trunk, or extremities. Multifocal lesions involve a wide variety of soft tissue locations and/or internal organs. Solitary soft tissue lesions are usually benign, but multifocal lesions with internal organ involvement may be fatal.
PATHOLOGIC FEATURES. IM may be well circumscribed or poorly demarcated. Microscopically, it contains two components in variable amounts: a highly vascular, cellular, Hemangiopericytoma (HPC)-like core and a smooth muscle–like myofibroblastic shell, the latter arranged in fascicles or whorls. Sometimes, IM can be quite cellular and mimic infantile fibrosarcoma. Mitoses (up to 10 per high-power field [hpf]) and intravascular tumor extension may be seen but do not influence outcome.17
ANCILLARY STUDIES. The myofibroblasts of IM show positive expression of SMA and variable expression of desmin, S100, Epithelial membrane antigen (EMA), and keratin. The spindle cells are negative for CD34, separating it from classic HPC/solitary fibrous tumor and from dermatofibrosarcoma protuberans of older children and adults.
Calcifying Aponeurotic Fibroma
CLINICAL FEATURES. Calcifying aponeurotic fibroma (CAF) is a fibromatosis affecting hands and feet of children. Its recurrence rate is about 50% after incomplete excision.18
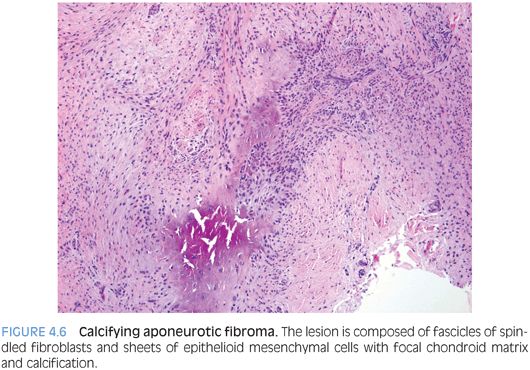
PATHOLOGIC FEATURES. CAF is usually a small lesion (mean size 2 cm) forming a firm, fibrous, infiltrative, gritty nodule. Histologically, CAF is composed of spindled fibroblasts and epithelioid mesenchymal cells with focal chondroid matrix and calcification (Fig. 4.6). Cellularity and amount of chondroid/calcified foci vary. Mitoses may be found, usually less than 2 per hpf. With time, the cellularity is reduced and chondroid foci are replaced by granular calcification surrounded by plump epithelioid fibroblasts and osteoclastic giant cells. Older tumors are hypocellular, composed predominantly of dense collagen with occasional fascicles of cellular fibroblasts and focal calcification.
ANCILLARY STUDIES. Ancillary study is usually not informative.
Inflammatory Myofibroblastic Tumor
CLINICAL FEATURES. Inflammatory myofibroblastic tumor (IMT) is a true neoplasm with intermediate biologic potential. It tends to occur in children, adolescents, and young adults, with a mean age of 10 years and sometimes with fever, weight loss, and/or anemia. IMT has a potential for local recurrence and metastasis.19
PATHOLOGIC FEATURES. Grossly, IMT is circumscribed but not encapsulated. The cut surface is white to tan, with a whorled, fleshy, or myxoid appearance. Histologically, IMT contains myofibroblastic and fibroblastic spindle cells and inflammatory infiltrates composed of lymphocytes and plasma cells (Fig. 4.7). The inflammation may obscure the myofibroblastic proliferation and abundant blood vessels may resemble HPC.

ANCILLARY STUDIES. IMT shows variable expression of SMA, muscle specific actin (MSA), desmin, and sometimes focal cytokeratin. Most cases (50% to 60%) express Anaplastic lymphoma kinase (ALK) in a cytoplasmic or nuclear membrane pattern; ALK expression occurs in other neoplasms and must be interpreted in context of other features. It may be a favorable prognostic indicator.19
Low-Grade Fibromyxoid Sarcoma
CLINICAL FEATURES. In pediatrics, low-grade fibromyxoid sarcoma (LGFMS) usually occurs in adolescents as a slow-growing, painless, infiltrative deep soft tissue mass that most commonly originates in the lower extremities. In children, it has a predilection to the head and neck and more superficial locations. It exhibits a deceptively benign fibromyxoid appearance but recurs frequently and occasionally metastasizes.
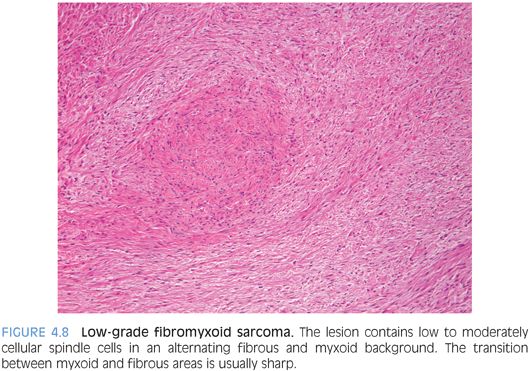
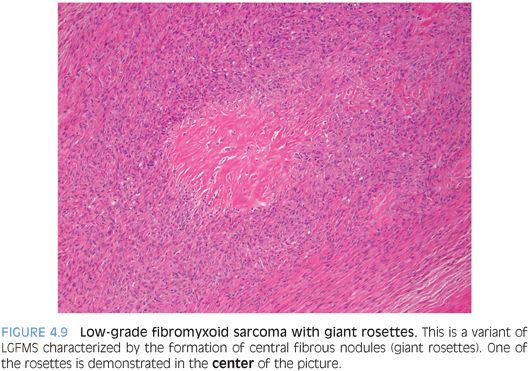
PATHOLOGIC FEATURES. Grossly, LGFMS is a deceptively well-circumscribed, 2- to 8-cm mass with a firm, white cut surface. Histologically, it contains low to moderately cellular spindle cells in an alternating fibrous and myxoid background, with a linear or whorled growth pattern. Myxoid areas often contain prominent curvilinear vessels with increased perivascular cellularity. There is a sharp zonation of myxoid and fibrous areas, but this transition may be gradual (Fig. 4.8). Hyalinizing spindle cell tumor with giant rosettes is a variant of LGFMS, characterized by containing large rosette-like structures with central collagen (Fig. 4.9). Uncommon findings include increased cellularity, pleomorphism, and epithelioid cells in a densely collagenized stroma mimicking sclerosing epithelioid fibrosarcoma, with which it appears to show biologic identity.
ANCILLARY STUDIES. LGFMS shows focal positivity for SMA and EMA, and rarely for desmin, CD34, and cytokeratin. Break-apart FISH for the FUS gene on chromosome 16 may assist in diagnosis.20 However, FUS is also disrupted in myxoid liposarcoma, so that the morphologic features of the tumor should be considered.
Infantile Fibrosarcoma
CLINICAL FEATURES. Infantile fibrosarcoma (IF) usually occurs in infants, and one-half are congenital. It chiefly affects the extremities but can present in unexpected locations. It usually forms a painless, rapid-growing mass in the subcutaneous and deep soft tissue, often reaching a huge size and infiltrating subcutaneous fat, muscle, fascia, and even bone. However, this tumor generally has a good clinical outcome.3
PATHOLOGIC FEATURES. IF forms a poorly circumscribed, invasive mass with a fibrous cut surface, histologically composed of hypercellular spindle cells arranged in interlacing fascicles and often accompanied by focal intercellular collagen. The tumor cells are uniform and plump with brisk mitoses. Like IM and IMT, some tumors show a pericytomatous pattern.
ANCILLARY STUDIES. IF expresses SMA but not desmin or CD34. It typically contains a characteristic TEL-NTRK3 fusion that differentiates it from other soft tissue neoplasms.21
MYOGENOUS TUMORS
Rhabdomyoma
Rhabdomyomas (RMs) are rare benign tumors with skeletal muscle differentiation, divided into two groups—cardiac and extracardiac. The latter is further subclassified into adult, fetal, and genital types.
CLINICAL FEATURES. Adult and genital RMs are nearly always found in adults and are not further discussed. Fetal RM is a rare tumor usually arising in the head and neck of infants and young children. Unlike rhabdomyosarcoma, fetal RM is usually superficially located in the subcutis or submucosa rather than in the muscle. Occasionally, it is associated with nevoid basal cell carcinoma syndrome or tuberous sclerosis.
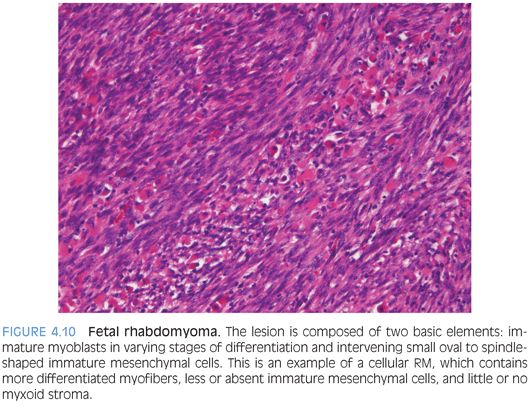
PATHOLOGIC FEATURES. Fetal RMs form well-circumscribed 2- to 6-cm masses, but larger lesions may occur. Mucosal tumors are generally polypoid. Histologically, they are composed of two basic elements: immature myoblasts in varying stages of differentiation and intervening small oval to spindle-shaped immature mesenchymal cells resembling fetal skeletal muscle. Myxoid and intermediate subtypes probably correspond to the duration of the lesions. The myxoid type contains conspicuous immature mesenchymal cells and myotubes within an abundant myxoid matrix. The intermediate type contains more differentiated myofibers, less or absent immature mesenchymal cells, and little or no myxoid stroma (Fig. 4.10). Myofibroblasts may be present. Cross-striations may not be easily found, especially in myxoid tumors, but myoid differentiation always can be demonstrated by special stains. Mitotic figures are rare or absent in both subtypes. Unusual features include focal infiltrative growth, increased mitoses (up to 14 per 50 hpf), focal necrosis, and mild to moderate nuclear pleomorphism. Marked nuclear atypia, anaplasia, and cambium layer should be absent.