Shock in the Gynecologic Patient
Addison K. May
DEFINITIONS
Categories of the etiologies of shock—
Hypovolemic
Blood loss, dehydration, and diarrhea
Distributive
Sepsis, endocrine, spinal, and anaphylaxis
Obstructive
PE, tamponade, pericarditis, and pulmonary hypertension
Cardiogenic
MI, myopathy, and valvular lesions
Classification of the severity of hemorrhagic shock—
Class I—up to 15% of blood volume
Compensation maintains normal cardiac output and blood pressure
Class II—15% to 30% of blood volume
Compensation cannot maintain normal cardiac output, but systolic blood pressure is maintained; pulse pressure is narrowed (compensated shock)
Class III—30% to 40% of blood volume
Cardiac output and blood pressure are decreased; significant tachycardia (uncompensated shock)
Class IV—greater than 40% blood loss
Cardiac output and blood pressure are profoundly decreased; very significant tachycardia
Compensated shock—A state of global, inadequate organ perfusion, and oxygen delivery with normal blood pressure.
Cytopathic hypoxia—A state of mitochondrial injury and dysfunction resulting from hypoxia and oxidative stress where oxidative phosphorylation is interrupted, thus mandating ATP generation through lactate generation.
Sepsis—The presence of SIRS due to an infectious etiology.
Septic shock—Sepsis advanced to the point of distributive shock.
Severe sepsis—When sepsis has progressed to a severity to manifest organ dysfunction such as delirium, acute lung injury, or acute kidney injury.
Severe SIRS—When SIRS has progressed to a severity to manifest organ dysfunction such as delirium, acute lung injury, or acute kidney injury.
Severe SIRS with shock—SIRS advanced to the point of distributive shock.
Shock—A state of global inadequate organ perfusion and oxygen delivery.
Systemic inflammatory response syndrome (SIRS)—Global activation of the inflammatory cascade, by a variety of stimuli, manifest by two or more of the following:
Hyper- or hypothermia (>38°C or <036°C)
Tachycardia (heart rate >90)
Increased minute ventilation (respiratory rate >20)
Leukocytosis or leukopenia (WBC > 12,000 or <4,000, or >10% bands)
Shock is defined as a state of global inadequate organ perfusion and may arise from a variety of diverse etiologies. Regardless of the etiology, the shock state will contribute to oxidative stress, cellular injury, and systemic activation of the inflammatory cascade (SIRS) and potentiate or lead to multisystem organ dysfunction syndrome (MODS). Both uncompensated and compensated shock will contribute to cellular changes that potentiate the severity of shock itself. An understanding of the physiologic derangements introduced by shock as well as the therapeutic interventions to mitigate the various etiologies of shock is required to optimally manage patients with this condition. In the chapter to follow, the etiologies, the physiologic cellular and organ derangements caused by the shock state, the interaction with the inflammation and organ injury, and the therapeutic interventions to target each are discussed.
DEFINITION AND ETIOLOGIES OF SHOCK
Shock is defined as a state of systemic or global inadequate organ perfusion and may be caused by a variety of insults. Most commonly, shock is recognized by hypotension, defining uncompensated shock. However, inadequate organ perfusion can occur with relatively normal vital signs, as occurs in compensated shock. Recognition that shock may exist without significant alterations in hemodynamics is critical to minimizing cellular and organ injury that may be introduced by occult hypoperfusion.
The various etiologies of shock are most frequently classified into four different categories based upon the underlying pathophysiologic alteration: hypovolemic, cardiogenic, obstructive, and distributive. Common causes of hypovolemic shock include hemorrhage, dehydration, and diarrheal diseases. Cardiogenic shock may be caused by an acute myocardial infarction, valvular lesions, and myopathies induced by ischemia, viral diseases, and inflammatory conditions. Causes of obstructive shock include tension pneumothorax, cardiac tamponade, constrictive pericarditis, acute pulmonary embolism, and severe pulmonary hypertension. The various causes of distributive shock include sepsis, spinal cord injury with neurogenic shock, adrenal insufficiency, vasopressin deficiency, anaphylaxis, and ischemia/reperfusion. Distributive shock physiologically differs from the other three classifications of shock in that a decline in cardiac output is not universally present. In fact, due to the decrease in systemic vascular resistance, cardiac output is frequently increased unless an underlying cardiac dysfunction also exists.
Appropriate therapeutic interventions for the treatment of shock should be directed toward the underlying pathophysiologic defect, which varies between and within classifications. While shock may be induced by a single etiology, recognizing that multiple etiologies may coexist and that one type of shock may cause or exacerbate a second is crucial to decisions regarding appropriate therapy. Changes induced by shock and the interdependence that may exist are outlined below.
PHYSIOLOGIC RESPONSE
Response to Decreased Cardiac Output
The predominant response to decreased cardiac output is to preserve perfusion to the heart and brain. This acute physiologic response is best characterized and understood in the context of hemorrhagic shock. Additionally, compensated shock is most easily understood in acute hemorrhagic shock, although inadequate organ perfusion with normal hemodynamics clearly exists in other forms of shock.
Acute hemorrhagic shock may be classified into four classes (Table 12.1). In class I shock, less than 15% of blood volume (approximately 750 mL in a 70 kg individual) has been lost. At this degree of blood loss, an increase in cardiac contractility and heart rate can maintain cardiac output and thus organ perfusion. In class II hemorrhagic shock, 15% to 30% of blood volume (approximately up to 1,500 mL in a 70-kg individual) has been lost. At this magnitude of blood loss, the acute compensatory mechanisms of increased heart rate and contractility can no longer maintain cardiac output. To maintain perfusion pressure, vasoconstriction of peripheral and mesenteric vascular beds increases resistance to blood flow, maintaining systolic blood pressure and increasing diastolic pressure (narrowed pulse pressure). While systolic blood pressure is preserved, cardiac output and delivery are decreased. This is the state of compensated shock. Class III hemorrhagic shock is defined as blood loss at 30% to 40% (approximately >1,500 mL in a 70-kg individual) of total body blood volume. At this degree of blood loss, cardiac output is severely altered and vasoconstriction can no longer maintain systolic pressure. Thus, perfusion pressure of the brain and heart are both now altered. Mental status changes and obvious signs of shock ensue. Class IV hemorrhagic shock is defined as greater than 40% of blood volume. At this degree of shock, profound hypotension, tachycardia, and severely depressed level of consciousness are manifested.
TABLE 12.1 Hemorrhagic Shock: Estimated Blood Lossa Based on Patient’s Initial Presentation | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
It is important to recognize that greater than roughly 1,500 mL of blood in a 70-kg individual must be lost before compensatory mechanisms are overwhelmed and hypotension develops. Significant blood loss with associated hypoperfusion of vital organs may exist prior to the development of hypotension. Inadequate organ perfusion induces cellular insult, as outlined below, regardless of blood pressure and must be recognized and treated.
Cellular Biochemical Changes
Shock, or inadequate perfusion, creates hypoxia of the cells involved, which results in anaerobic rather than aerobic metabolism for cellular functions. This period of anaerobic metabolism produces “hypoxic priming” within the affected cells, characterized by a marked reduction in adenosine triphosphate (ATP) generation by the cell, an increase in oxidative stress, loss of antioxidant potential, and a buildup of acidic by-products, including lactic acid. The decrease in ATP production within hypoxic cells leads to a cascade of events including decreased ion exchange by the sodium-potassium adenosine triphosphatase pumps (Na+/K+-ATPase), calcium (Ca2+), cellular swelling, Ca2+ activation of phospholipases with activation of the arachidonic acid cascade, and generation of fatty acid reactive oxidant species. Additionally, cellular hypoxia leads to the ischemic generation of xanthine oxidase by irreversible proteolytic cleavage of xanthine dehydrogenase, the enzyme that normally catalyzes the metabolism of hypoxanthine (product of ATP utilization) to xanthine and subsequently uric acid via the generation of NADH from NAD+.
Reperfusion of cells following a period of hypoxic priming induces further oxidant injury and activation of the inflammatory cascade through several mechanisms. The xanthine oxidase enzyme, generated during hypoxia, catalyzes the conversion of hypoxanthine and oxygen to xanthine and subsequently uric acid and generating the oxidative species, superoxide (O2–). Additionally, hypoxia followed by reperfusion induces increased activity of nitric oxide synthetase and enhanced production of nitric oxide (NO), another oxidative species. NO and O2– interact to form yet another oxidative species, peroxynitrite (ONOO). These oxidative species generated by hypoxia and reperfusion directly injure mitochondria, proteins, chromosomes, and membrane structures. In severe shock states, oxidative stress can be greatly potentiated by cellular mitochondrial injury, a condition known as cytopathic hypoxia. Oxidative species, such as ONOO, can oxidize the components of the electron transport chain and open pores in the mitochondrial membrane, both contributing to mitochondrial dysfunction and loss of aerobic metabolism by the affected cells. The loss of mitochondrial function mandates anaerobic metabolic pathways by the affected cells, an ongoing reduction of ATP generation, thus further potentiating oxidative stress. The oxidative cellular injury introduced by shock potentiates oxidative injury produced by tissue trauma, infection, and sepsis as shown in Figure 12.1.
Global hypoxia and reperfusion directly activates the proinflammatory nuclear transcription factor NF-κB with increased production of cytokines (TNF, IL-1, IL-6, IL-8), chemokines, coagulation factors, adhesion molecules, and activation of tissue macrophages and neutrophils. The shock state also directly activates the innate immune system through the release of a variety of substances (referred to as “damage-associated molecular patterns” or DAMPs) that are recognized by tolllike receptors (particularly TLR4) expressed on endothelial cells, neutrophils, and tissue macrophages. Systemic activation of macrophages and neutrophils results in direct tissue damage and still further cellular oxidant injury. As in the case oxidant generation, activation of the proinflammatory state and the innate immune system by shock potentiates the activation of the inflammatory cascade caused by other insults such as tissue trauma, infection, and sepsis.
Shock and SIRS
The persistence of the shock state creates a nonlinear increase in oxidative injury and activation of the inflammatory cascade and innate immune system. Increasing severity and persistence of shock will produce systemic inflammatory response syndrome (SIRS) and organ dysfunction, culminating in MODS.
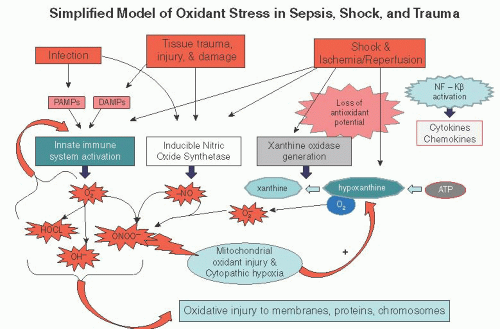 FIGURE 12.1 This simplified model of oxidant stress in sepsis, shock, and trauma demonstrates the overlapping nature of oxidant generation from these insults. Infection, tissue injury, and ischemia/reperfusion injury all activate the innate immune system via the molecular patterns recognized by toll-like receptors on immune cells which in turn produce several oxidative species. Additionally, nitric oxide production is up-regulated. Shock and ischemia/reperfusion injury may also irreversibly cleave xanthine dehydrogenase to xanthine oxidase, increasing the production of oxidant stress. Finally, significant cellular oxidative stress can induce mitochondrial injury and cytopathic hypoxia. PAMPs, pathogen-associated molecular patterns, DAMPs, damage-associated molecular patterns. Oxidative species: HOCL, hypochlorous acid; O2–, superoxide; NO, nitric oxide; OH–, hydroxide; ONOO–, peroxynitrite. |
Additionally, shock also creates and enhances vasodilation resulting in distributive shock. SIRS can result from or be exacerbated by shock. Activation of the inflammatory cascade and the subsequent proinflammatory state outlined above produces physiologic changes in temperature (hyperthermia, temperature > 38°C or less commonly hypothermia, temperature < 36°C), heart rate (tachycardia HR > 90), minute ventilation (respiratory rate > 20), and changes in white blood cell count (leukocytosis WBC > 12,000, bandemia > 10% bands, or less commonly leukopenia WBC < 4,000), with two or more of these changes defining SIRS. As the severity of this process progresses, organ dysfunction will be manifest, thus defining severe SIRS. The presence of SIRS and severe SIRS in which the etiology is infectious defines sepsis and severe sepsis, respectively. In critically ill patients, SIRS and sepsis are very common, occurring in roughly 75% to 80% of this population. However, SIRS is significantly more common than is sepsis, a fact that should be considered when making decisions regarding empiric antibiotics in response to fever and leukocytosis.
Vasodilation
During shock, vascular smooth muscle function is altered leading to vasodilation. Vascular smooth muscle function is directly altered by a loss of ATP production, decreased cytoplasmic calcium and decreased phosphorylation of myosin, decreasing smooth muscle contraction. The resulting vasodilation is propagated by the up-regulation of nitric oxide synthetase and subsequently increased nitric oxide production, resulting in even greater vascular smooth muscle relaxation. As a result, systemic vascular resistance falls and distributive (or vasodilatory) shock develops. Thus, a distributive shock state may develop secondary to other forms of shock. The vasodilated state may persist for days and may require prolonged vasoconstrictive agents to ensure adequate mean arterial pressure (MAP) to ensure tissue perfusion. Increasing severity and length of hypoperfusion increases the magnitude of this series of events. Thus, hypovolemic, obstructive, and cardiogenic shock may all initiate a state of distributive or vasodilatory shock following resuscitation. After correction of the original underlying physiologic defect (e.g., hemorrhagic shock), subsequent therapy may be required to correct the vasodilatory component rather than continuing pure volume replacement.
Additionally, tissue hypoxia and sepsis both may lead to defects in the production of vasopressin by the hypothalamus and cortisol by the adrenal glands. Additionally, the use of etomidate as an induction agent for endotracheal intubation is commonly associated with a period of adrenal insufficiency. Deficiencies of either vasopressin or cortisol will produce vasodilation and distributive shock that is refractory to inotropic support. These deficiencies will coexist with the distributive shock resulting from nitric oxide-induced vasodilation and may complicate management of other forms of shock.
Coagulopathy
While large volumes of blood loss may be sustained without the development of coagulopathy if normotension is maintained, coagulopathy develops at much a much smaller volume of blood loss if shock is present. Tissue hypoxia directly potentiates coagulopathy, and on-going shock will worsen derangements that are introduced by blood loss, hypothermia, and infection. Significant overlap of the coagulation system, the anticoagulant and fibrinolytic pathways, and the inflammatory system exists, and hypoperfusion and tissue hypoxia will potentiate alteration in both pro- and




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
