Rhabdomyosarcoma and Nonrhabdomyomatous Sarcomas
Dave R. Lal
Michael P. LaQuaglia
Dave R. Lal: Department of Surgery, Memorial Sloan-Kettering Cancer Center, New York, New York 10021.
Michael P. LaQuaglia: Department of Surgery, Weill-Cornell University Medical School, Memorial Sloan-Kettering Cancer Center, New York, New York 10021.
The study of soft-tissue malignant tumors arising from mesenchyme has been revolutionized by developments in molecular embryology, cytogenetics, and gene cloning. Advances in these fields have in some cases eliminated total reliance on light microscopic histology and immunohistochemistry for correct diagnosis. As knowledge evolves, the prospect of molecular staging and use of genetic analysis for determination of risk status will become a reality.
This chapter organizes the soft-tissue sarcomas by cell of origin. It begins with a discussion of relevant scientific developments, which is followed by an analysis of diagnoses, staging, and treatment. Finally, future developments in treatment are explored.
CLINICAL PROBLEM
Soft-tissue sarcomas are the sixth most common malignancy of childhood, with rhabdomyosarcoma by far the most frequent. In the United States, 850 to 900 pediatric soft-tissue sarcomas are diagnosed each year, with about 350 being rhabdomyosarcomas (RMSs) (1). The yearly incidence of RMS is about 4.3 per 1 million children younger than 20 years of age (1). In comparison, other sarcomas such as fibrosarcoma, malignant fibrous histiocytoma, tenosynovial sarcoma, neurofibrosarcoma, primitive neuroectodermal tumor (PNET), and leiomyosarcoma each occur at an incidence between 0.3 and 0.8 cases per million children per year (1,2,3,4). The incidence of desmoplastic small round cell tumors (DSRCTs), malignant rhabdoid tumors, alveolar soft parts sarcoma, hemangiopericytoma, myofibrosarcoma, epithelioid sarcoma, and liposarcomas is nearly zero in childhood, although small series have been reported. The incidence of osteosarcoma is 2.82 per 1 million children, and that of Ewing’s sarcoma is 2.76 per 1 million children, but these are primarily lesions arising from bone. Extraosseous osteosarcoma and Ewing’s sarcoma are rare. Each histopathologic subtype has characteristic biologic, clinical, and prognostic parameters that influence treatment. Figure 37-1 shows the relative percentages of sarcomas presenting to a major cancer center during a 10-year period. Figure 37-2 is a breakdown of rare soft-tissue sarcomas by histologic subtype. Figure 37-3 depicts the age distributions of soft-tissue sarcoma at diagnosis. The principal therapeutic problem in all soft-tissue sarcomas is matching treatment intensity to prognostic risk. Important to this consideration are underlying biologic factors and clinical prognostic variables.
RHABDOMYOSARCOMA
History
Since the mid-1960s, improved survival with decreased morbidity has been accomplished in the treatment of children with rhabdomyosarcoma. The Intergroup Rhabdomyosarcoma Study Group (IRSG) has been responsible for much of the advancements in treatment, improved survival, and decreased morbidity. The IRSG was first established in 1972. At that time surgery was the main form of treatment, and chemotherapy had just started to emerge as a treatment modality. Due to the relative paucity of RMSs seen at any one institution, meaningful clinical trials were rarely performed. The IRSG mission has been to enroll all children in North America with RMS into clinical trials, with its main goal of improving survival while decreasing patient morbidity. More than 80% of North American children diagnosed with RMS have been enrolled in one of four completed IRSG prospective randomized trials. The IRSG has fostered a multidisciplinary approach to rhabdomyosarcoma, including surgeons, oncologists, and radiation oncologists. Since the inception of the IRSG, the overall survival for patients with RMS has improved from 25% to more than 70%. The IRSG (now called the Children’s Oncology Group Soft-tissue Sarcoma Committee) is now accruing patients for Intergroup Rhabdomyosarcoma Study V (IRS-V). The goals of this study are to evaluate the use of new chemotherapeutic regimens, abolish the use of radiation therapy for patients with low risk of recurrence (completely resected, group I embryonal RMS), and search for molecular markers to help predict biologic activity and response to treatment (5).
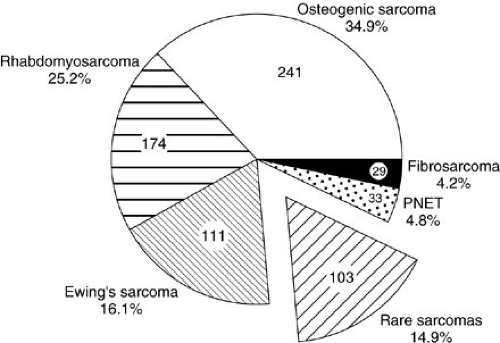 FIGURE 37-1. Relative proportions of childhood sarcomas diagnosed at the Memorial Sloan-Kettering Cancer Center during a 10-year period. PNET, peripheral primitive neuroectodermal tumor. |
Epidemiology
RMS is the most common soft-tissue sarcoma of childhood. It accounts for 5% to 8% of cases of childhood cancer and for about 70% of the sarcomas. Since 1975, the yearly incidence of RMS has remained stable at approximately 4 per million (1). The incidence in African American females is about one-half that in white children. RMS of the bladder, vagina, and head and neck region usually occurs in infants and small children; extremity and truncal lesions predominantly occur in later childhood and adolescence (4).
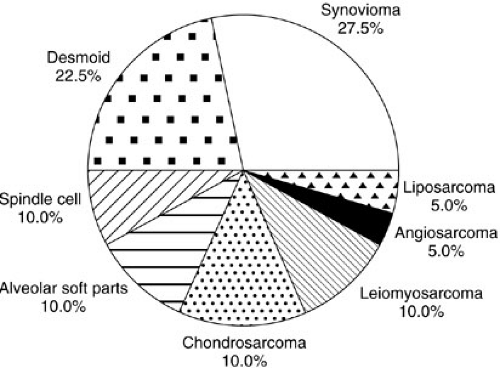 FIGURE 37-2. Relative proportions of rare childhood soft-tissue tumors observed during a 10-year period. |
Pathology
RMSs are malignant tumors arising from the ubiquitously distributed primitive mesenchyme found in the fetus. These tumors display characteristics of striated muscle, including immunohistochemical expression of skeletal muscle myosin and actin, desmin, myoglobin, and Z-band protein. Electron microscopy may show actin and myosin bundles or Z-band material. Expression of the DNA-binding proteins MYOD1 and myogenin have shown MYOD1 to act as a linkage marker, while myogenin acts as a differentiation marker (6,7,8). These characteristics allow the differentiation of RMS from other common pediatric tumors, including lymphoma, primitive neuroectodermal tumor, Ewing’s sarcoma, and neuroblastoma.
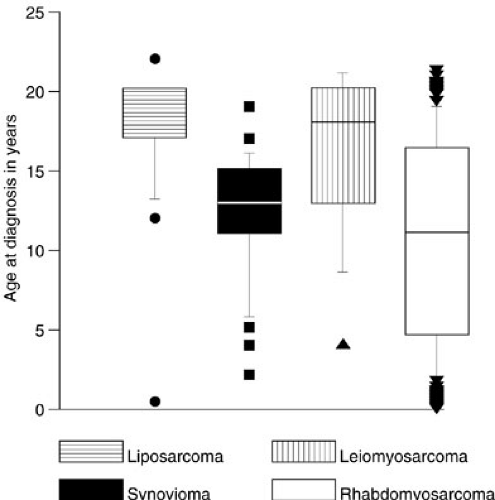 FIGURE 37-3. Age distributions at diagnosis of four types of soft-tissue sarcomas diagnosed in childhood and adolescence. |
Weber first published a description of RMS in 1854. In 1958, Horn published a series of 39 patients describing their pathologic characteristics and creating four main subtypes of RMS based on histolopathology: embryonal, botryoid (subtype of embryonal), alveolar, and pleomorphic (9). This classification system was modified in 1995 into the International Classification of Rhabdomyosarcoma (ICR) shown in Table 37-1. The ICR was created by an international consensus of pathologists reviewing 800 cases of RMS from the IRS-II. The advantage of this classification is its reproducibility between pathologist and improved prediction of outcome (10).
In the most recent IRS-IV, embryonal tumors represented the most common (74%) histologic subtype of rhabdomyosarcoma diagnosed in children (11). A breakdown of histology for all patients enrolled in IRS-I to -IV is displayed in Fig. 37-4. Embryonal RMS is seen more commonly in younger children and in those with head and neck and urinary tract tumors. Microscopic characteristics of embryonal RMS include ribbon-shaped cells with elongated nuclei and loose stroma, as illustrated in Fig. 37-5. Botryoid and spindle tumors represent a subtype of embryonal RMS with a markedly improved survival. Botryoid tumors grow into a hollow space (i.e., vagina, bladder, nasopharynx), so they develop a characteristic grapelike gross morphology. Microscopically, these tumors form a dense layer of neoplastic cells beneath the epithelium known as the cambial layer. Spindle tumors are characterized by nearly uniform spindle-shaped cells with low cellularity and storiform patterns; these tumors are more common in paratesticular RMS. Alveolar tumors are so named because of a resemblance to the microscopic structure of the lung (Fig. 37-6), with thin fibrous septae lined by neoplastic cells. RMSs are identified as alveolar if any alveolar elements are found in the tumors. These tumors are more common in older children and those with trunk or extremity RMS. Undifferentiated RMS contains primitive noncommitted mesenchymal cells and is a diagnosis of exclusion. For all RMS cell types, sites of occurrence include the head and neck (orbit, infratemporal fossa); genitourinary tract, including perineum and perianal area; extremities; trunk (chest wall, paraspinal area); retroperitoneum; gastrointestinal tract; biliary tract; and heart. Figure 37-7 displays the location of primary RMS found in IRS-I to -IV.
TABLE 37-1 International Classification of Rhabdomyosarcoma. | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
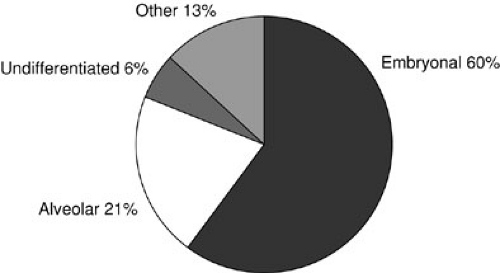 FIGURE 37-4. Distributions of histology in patients enrolled in Intergroup Rhabdomyosarcoma Study I to IV. |
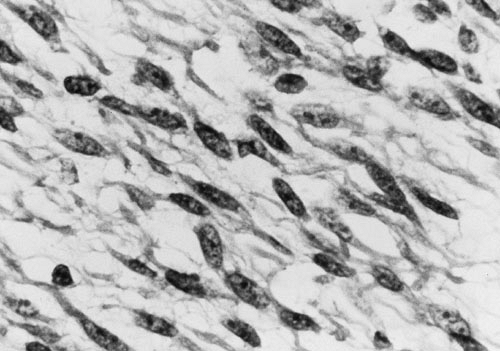 FIGURE 37-5. Photomicrograph of an embryonal rhabdomyosarcoma specimen. |
Genetics
A majority of RMS are sporadic in nature. The incidence of inheritable RMS is unknown but has been estimated at between 7% and 33% (12). RMS has been associated with three autosomal dominantly inherited disorders: Li-Fraumeni syndrome, neurofibromatosis type I, and Beckwith-Wiedemann syndrome. Li-Fraumeni syndrome patients have a propensity for RMS, premenopausal breast cancer, osteosarcoma, brain tumors, adrenal cortical carcinomas, and acute leukemia. A germline mutation in p53 has been associated with Li-Fraumeni syndrome. Neurofibromatosis type 1 patients have mutations in the tumor suppressor gene NF1. Beckwith-Wiedemann syndrome is characterized by macroglossia, organomegaly, and macrosomia. Beckwith-Wiedemann syndrome patients have been found to have alterations at the 11p15 chromosomal locus, which harbors the gene for IGF-II (13). Coincidentally, this locus is also altered in embryonal RMS.
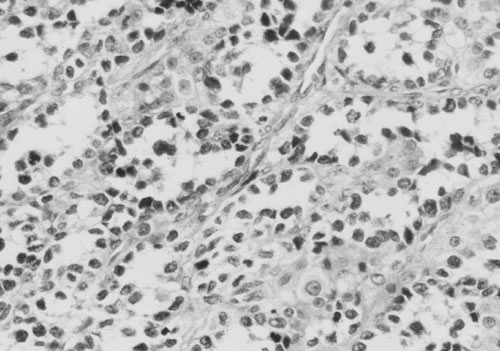 FIGURE 37-6. Photomicrograph of an alveolar rhabdomyosarcoma specimen illustrating the lunglike cytoarchitecture. |
Presentation and Diagnostic Evaluation
The incidence of RMS is biphasic, with one peak in infancy followed by a second in adolescence. Presentation is site dependent. Head and neck lesions can cause facial or cervical swelling and associated pain or skin discoloration. Sinusitis or middle ear infections can occur because the tumor blocks the normal drainage from these sites (i.e., sinusoidal ostia, eustachian tubes). Epistaxis, proptosis, or cranial nerve palsies may also be evident in head and
neck lesions. Genitourinary tumors present with gross or microscopic hematuria, a suprapubic mass, or urinary tract infection or obstruction. Vaginal and cervical primary tumors often prolapse through the vaginal orifice as a friable polypoid mass and may hemorrhage. Paratesticular lesions are most often diagnosed in adolescence as a hard mass above the testis. Although difficult to always demonstrate, physical exam reveals the mass and testis to be separable. Extremity tumors present as painless or painful expanding masses, and there may be an associated limp or overlying skin change. Local bony invasion may result in pathologic fractures.
neck lesions. Genitourinary tumors present with gross or microscopic hematuria, a suprapubic mass, or urinary tract infection or obstruction. Vaginal and cervical primary tumors often prolapse through the vaginal orifice as a friable polypoid mass and may hemorrhage. Paratesticular lesions are most often diagnosed in adolescence as a hard mass above the testis. Although difficult to always demonstrate, physical exam reveals the mass and testis to be separable. Extremity tumors present as painless or painful expanding masses, and there may be an associated limp or overlying skin change. Local bony invasion may result in pathologic fractures.
 FIGURE 37-7. Location of primary rhabdomyosarcoma at diagnosis in patients enrolled in the Intergroup Rhabdomyosarcoma Studies I to IV. |
Rational management depends on a thorough pretreatment workup that defines completely local tumor extent, as well as evaluating regional and distant sites of metastases. Table 37-2 lists the standard workup for pediatric sarcoma patients. An extensive history, including family history of breast cancer or other forms of sarcoma, and thorough physical examination are elementary. Radiographic staging has been markedly facilitated by modern imaging methods. Even greater progress in this area is promised by efforts in the field of positron emission scanning, which allows determination of the metabolic activity of a tumor mass. The T2-weighted magnetic resonance image appearance of a large RMS arising in the buttock of a 3-year-old is shown in Fig. 37-8.
Staging
An evolution has occurred in sarcoma staging since the mid-1990s, with most groups adopting the modified tumor nodes metastasis (TNM) system defined by the IRSG for use in IRS-IV (14). The TNM-based schema attempts to divide stage into definable clinical components that are assessed prior to any therapeutic intervention. This system categorizes site of origin, invasiveness (confined to site of origin), size of lesion (less than or greater than 5 cm), nodal disease (positive or negative), and metastatic status into a stage that has been shown to be predictive of patient outcome. In examining data from IRS-II, anatomic location of the primary tumor was shown to have prognostic significance (15). Therefore, the site of the primary tumor was incorporated into this staging system (Table 37-3). Tumors located in the orbit, head and neck (excluding parameningeal), and genitourinary (excluding bladder and prostate) locations have better prognosis, and this is reflected in their stage. In contrast, tumors of the extremity, bladder, prostate, trunk, retroperitoneum, abdominal, or cranial parameningeal location are more
aggressive and are upstaged. Examining data from IRS-IV, patients with metastatic disease had a higher percentage of tumors greater than 5 cm (82% vs. 51%), invasive tumors (91% vs. 49%), positive regional lymph nodes (57% vs. 16%), and alveolar histology as compared with patients presenting without metastatic disease (11,16). The IRSG TNM staging system and its approximate correlation with IRSG surgicopathologic clinical group system are listed in Table 37-4. The IRSG surgicopathalogic clinical group system allows a second classification based on surgical and histologic findings of the primary tumor and regional lymph nodes after resection. Figure 37-9 displays the distribution of patients enrolled in IRS-I to -IV by clinical group.
aggressive and are upstaged. Examining data from IRS-IV, patients with metastatic disease had a higher percentage of tumors greater than 5 cm (82% vs. 51%), invasive tumors (91% vs. 49%), positive regional lymph nodes (57% vs. 16%), and alveolar histology as compared with patients presenting without metastatic disease (11,16). The IRSG TNM staging system and its approximate correlation with IRSG surgicopathologic clinical group system are listed in Table 37-4. The IRSG surgicopathalogic clinical group system allows a second classification based on surgical and histologic findings of the primary tumor and regional lymph nodes after resection. Figure 37-9 displays the distribution of patients enrolled in IRS-I to -IV by clinical group.
TABLE 37-2 Diagnostic Evaluation for Suspected Rhabdomyosarcoma. | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
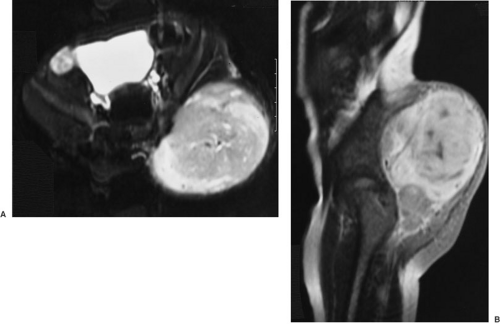 FIGURE 37-8. A: T2-weighted axial magnetic resonance image showing a large rhabdomyosarcoma of the left buttock in a 3-year-old child. The indistinct border suggest local tumor invasiveness. B: Sagittal view of the same tumor. |
TABLE 37-3 Site of Primary Rhabdomyosarcoma and Prognosis. | ||||||
|---|---|---|---|---|---|---|
| ||||||
Local tumor invasion is the consequence of cellular and molecular phenotypic properties, which are rapidly being defined by molecular analyses. The ability of cells to invade surrounding basement membranes depends on manufacture of a spectrum of lytic enzymes and on cellular motility. Furthermore, regional or distant metastases are not possible without initial cellular invasion. The gross invasion assessed for by TNM staging is a rough measure of these cellular events. In this sense, the TNM system begins to relate macroscopic tumor behavior to the properties and interactions of individual tumor cells. Future directions include more precise definitions of the process of tumor cell invasion. Assays of collagenolytic activity and growth and invasion in nude mice are intermediate steps in this regard. Eventually, genetic determinants of invasion and metastasis that are the final determinants of prognosis will be identified.
A sarcoma is adequately staged when the following criteria are met:
The primary tumor pathology and histopathologic subtype have been determined by biopsy.
All components in the TNM staging system are known with reasonable certainty.
TABLE 37-4 Comparison of Two Staging Systems.
Stage
Primary Site
Tumor Invasiveness
Tumor Size
Nodal Status
Metastasis
Group
Criteria
IRSG TNM-based pretreatment staging systema
IRSG surgicopathologic clinical groups
I
Orbit
Head and neck (excluding parameningeal)
Genitourinary (excluding bladder and prostate)
T1 or T2
a or b
N0, N1, or Nx
M0
I
A
B
Localized completely resected disease
Confined to organ or muscle of origin
Infiltration outside organ or muscle of origin, but no regional nodal involvement
II
Bladder or prostate
Extremity
Head and neck parameningeal
Other unfavorable sites
T1 or T2
a
No or Nx
M0
II
A
B
C
Compromised or regional resection
Gross resection with microscopic residual
Regional disease, completely resected, but nodes may be involved, or there is extension into adjacent organs
Regional disease, nodes involved, microscopic residual disease after resection
III
Bladder or prostate
Extremity
Head and neck parameningeal
Other unfavorable sites
T1 or T2
T1 or T2
a
b
N1
N0, N1,
or Nx
M0
M0
III
Incomplete resection or biopsy alone with gross residual disease
IV
All
T1 or T2
a or b
N0 or N1
M1
IV
Distant metastases at diagnosis
aT1, confined to anatomic site of origin; T2, extension beyond site of origin; a ≤ 5 cm; b > 5 cm; N0, nodes not clinically involved; N1, nodes clinically positive; Nx, unknown clinical nodal status; M0, no distant metastasis; M1, metastasis present.
Reprinted from Lawrence W, Anderson JR, Gehan EA, et al. Pretreatment TNM staging of childhood rhabdomyosarcoma: a report of the Intergroup Rhabdomyosarcoma Study Group. Children’s Cancer Study Group. Pediatric Oncology Group. Cancer 1997;80:1168, with permission.
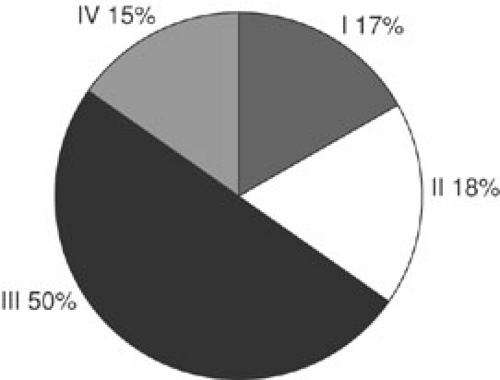
FIGURE 37-9. Distribution by clinical group in patients enrolled in the Intergroup Rhabdomyosarcoma Studies I to IV.
Biopsy should be performed on metastatic regional lymph nodes, but extensive nodal dissections are not indicated except in extremity and paratesticular RMS. Unique to extremity RMS is the marked difference in 5-year failure-free survival, 33% versus 68% (p < 0.001) in regional lymph node positive disease versus regional node negative disease, respectively (17). In IRS-IV, 17% of nodes that were clinically negative were found to have microscopic disease upon excision and pathologic examination (17). Although regional lymph node dissection does not improve survival, it is essential for proper staging and treatment of these high-risk children. Sentinel lymph node biopsy has recently been described in RMS as a method to assess the axillary and femoral nodes without the morbidity of a formal dissection (18,19). More extensive trials are required to determine its validity in pediatric patients with RMS.
In paratesticular RMS, a limited dissection of ipsilateral, periaortic nodes had been used as a determinant of the need for nodal irradiation and intensified chemotherapy. IRS-IV staged the periaortic nodes by computed tomography (CT) scan only. Nodes larger than 2 cm were considered positive and received adjuvant therapy. Analysis of outcomes revealed that CT scan determination of nodal status did not affect survival rates in children younger than 10 years old. However with this protocol, children older than age 10 had higher relapse rates and decreased survival. Therefore, the standard treatment for children ages 10 or older with paratesticular RMS is ipsilateral periaortic nodal dissection (20). Many European groups do not perform nodal biopsy for paratesticular RMS and instead treat patients older than age 10 or with tumors greater than 5 cm with intensified alkylating agent chemotherapy (21).
Small, accessible primary lesions should undergo excisional biopsy. The surgeon should attempt to obtain a clear microscopic margin. Larger infiltrating lesions or those whose removal causes debilitation or deformity (i.e., amputation or cystectomy) should undergo limited incisional or endoscopic biopsy. It is acceptable to perform transperineal needle-core biopsies for bladder neck or perineal primary tumors. Enough biopsy material for light and electron microscopic analysis, immunohistochemistry, and cytogenetics should be obtained. It is preferable to freeze extra tissue to facilitate further specialized studies. Even though such investigations do not have direct, short-term benefit for the patient, they are crucial in expanding knowledge concerning RMS. Tissue should be frozen in liquid nitrogen as soon as possible after biopsy.
Treatment
The modern treatment of RMS is multidisciplinary and includes multiagent chemotherapy, judicious resection, and radiotherapy. Topotecan and Irinotecan, topoisomerase I inhibitors, have shown promising results for children with metastatic RMS. Response rates have been reported between 46% and 86%, and both have been incorporated into the treatment algorithm for IRS-V (22,23). The intensity of chemotherapy should be tailored to the risk of subsequent relapse, which is a function of TNM stage. In general, agents are combined to limit drug resistance while attaining a synergistic antitumor effect. A major surgical responsibility during chemotherapy is maintenance of adequate vascular access both for administration of medications and for blood sampling. An external or implanted vascular access device can usually be inserted at the time of diagnostic biopsy.
Surgical resection of the primary tumor was the mainstay of treatment in the mid-1960s, but resulted in overall survival rates in the range of 20%. This improved to 50% with the addition of chemotherapy, resulting in a period when the role of tumor resection in RMS was questioned. It is difficult to prove that surgical removal of the primary tumor alters survival probability. A major reason is that a significant proportion of patients have distant metastases at the time of presentation, and this so profoundly influences survival that the effect of other variables is obscured. In addition, most tumors that can be readily removed without serious disfigurement or disability are resected. Thus, the effect of leaving a tumor in situ cannot readily be evaluated. With present survival rates, it would be unethical to randomize patients with nonmetastatic, resectable tumors to a nonsurgical arm. It is recommended that complete resection of primary tumors should be undertaken either before chemotherapy for small noninvasive lesions or after documented response with more formidable primary tumors. In certain situations in which chemotherapy results in complete or very good tumor regression, external-beam radiotherapy can be used as a primary means of local control. Even in these circumstances, biopsy is important to document complete tumor eradication. Debilitating or disfiguring surgery should only be performed if residual tumor is present after both chemotherapy and therapeutic radiotherapy. Amputation for extremity RMSs does not enhance cure and should be performed only when lesions are bulky, invade bone or neurovascular structures, or are recurrent. Similarly, radical cystectomy, hysterectomy/vaginectomy, and eye enucleation are reserved for situations in which complete tumor eradication has not been accomplished by chemotherapy and external-beam radiotherapy. The guiding principle of RMS surgery is complete tumor resection; however, removal of large amounts of adjacent normal tissue (i.e., muscle group resection, amputation) does not, in itself, affect outcome.
Controversy exists regarding treatment of patients with residual microscopic disease after excision. Analyzing outcomes of group 2 patients from IRS-I to -IV identified a subgroup of patients that had improved survival and lower relapse rates. Embryonal histology, favorable primary site, and subgroup IIa or IIb were identified on multivariate analysis to confer improved 5-year failure-free survival (88% to 93% vs. 38% to 78% in all other patients) (24). It was hypothesized that group II patients who do not possess these characteristics associated with improved survival may benefit from reexcision. Hays and associates (25) showed that patients with group II truncal or extremity RMS had improved 3-year survival after undergoing reexcision of the primary tumor with negative margins (91% 3-year survival vs. 74% in nonreexcision group). By reexcising the primary tumor, these patients had improved survival and were downstaged to group I, thus decreasing the intensity of adjuvant therapy.
External-beam radiotherapy of the primary site or involved regional nodal echelons has contributed to locoregional control. Clinical data suggest that control of
microscopic RMS can be accomplished with doses of 4,000 cGy, whereas gross deposits require more than 5,000 cGy for sterilization (26). IRS-IV sought to improve local/regional control rates while minimizing toxicity. Group III (incomplete resection with gross residual tumor) patients were randomized to conventionally fractionated radiotherapy (daily 180 cGy for a total dose of 5,040 cGy) versus hyperfractionated radiotherapy (twice-daily 110 cGy for a total dose of 5,940 cGy) (27). No difference in failure-free survival or overall survival was seen. However, the hyperfractionated group suffered from more mucositis, skin reactions, and nausea/vomiting (27). Conventionally fractionated radiotherapy has been adopted for use in IRS-V. Treatment typically begins 9 weeks after neoadjuvant chemotherapy (exception is parameningeal tumors in which treatment is started immediately) and continues for 5 to 6 weeks. The radiation field encompasses the entire area that the tumor occupied prior to surgery or chemotherapy along with a 2-cm margin.
microscopic RMS can be accomplished with doses of 4,000 cGy, whereas gross deposits require more than 5,000 cGy for sterilization (26). IRS-IV sought to improve local/regional control rates while minimizing toxicity. Group III (incomplete resection with gross residual tumor) patients were randomized to conventionally fractionated radiotherapy (daily 180 cGy for a total dose of 5,040 cGy) versus hyperfractionated radiotherapy (twice-daily 110 cGy for a total dose of 5,940 cGy) (27). No difference in failure-free survival or overall survival was seen. However, the hyperfractionated group suffered from more mucositis, skin reactions, and nausea/vomiting (27). Conventionally fractionated radiotherapy has been adopted for use in IRS-V. Treatment typically begins 9 weeks after neoadjuvant chemotherapy (exception is parameningeal tumors in which treatment is started immediately) and continues for 5 to 6 weeks. The radiation field encompasses the entire area that the tumor occupied prior to surgery or chemotherapy along with a 2-cm margin.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


