Renal Tumors
Robert C. Shamberger
Department of Surgery, Harvard Medical School, Children’s Hospital, Boston, Massachusetts 02115.
Renal tumors are the second most common abdominal tumor presenting in infants and children. They represent a broad spectrum of disease from the benign to an extremely malignant tumor. Advances in their management have included several landmark events, such as the first administration of chemotherapy for a solid tumor and early administration of chemotherapy in an “adjuvant” setting, long before it was accepted for many tumors occurring in adults. Much of what we know about these tumors today has resulted from two cooperative group organizations, the National Wilms’ Tumor Study (NWTS) group and the Sociètè Internationale d’ Oncologie Pèdiatrique (SIOP). Together, they have performed multiple randomized therapeutic trials that have established the basis for how these tumors are treated today. Moreover, these studies have also produced the current pathologic classification and staging of these tumors, which could never have been established without multiinstitutional participation because of the relative rarity of these tumors. In this chapter the early history of Wilms’ tumor is presented, followed by a discussion of the etiologic factors in tumor formation, pathologic subtypes and premalignant syndromes, and treatment algorithm for Wilms’ tumors and other tumors of the kidney.
HISTORY
William E. Ladd and Robert E. Gross described the surgical therapy of Wilms’ tumor, including preliminary ligation of the renal pedicle (1,2). They also stressed the need to remove the perirenal fat to include lymphatic extensions and to avoid rupture of the renal capsule, principles we continue to follow to this day.
Wilms’ tumor was the first malignancy in which the importance of adjuvant treatment of the tumor was recognized. From 1931 to 1939, survival from surgical resection alone, involving ligation of the renal pedicle before removal, was 32% at Children’s Hospital in Boston (3). Beginning in 1940, most of the patients received postoperative radiation to the renal fossa, which decreased the local recurrence rate, but did not significantly impact the frequency of pulmonary metastases or improve the long-term survival. The need for adjuvant treatment as perceived by Sidney Farber was based on the supposition that in the children with Wilms’ tumor who died, the tumor must have metastasized already at the time of discovery of the primary tumor, although no evidence of spread was available (3). Actinomycin D was the first active agent identified for the treatment of Wilms’ tumor. Of the 53 patients who had no demonstrable metastases on admission treated with combined therapy of surgery, local radiation, and actinomycin D from 1957 to 1964, an 89% 2-year disease-free survival was reported, a very reasonable survival even today (3). In patients with metastases identified at presentation, 18 of 31 (58%) were alive and free of disease longer than 2 years later. Subsequently, vincristine sulfate was identified as an active agent in Wilms’ tumor and was added to the standard therapy (4).
WILMS’ TUMOR INCIDENCE AND ETIOLOGY
Wilms’ tumor is the most frequent tumor of the kidney in infants and children. Its incidence is 7.6 cases for every 1 million children younger than 15 years of age or 1 case per 10,000 infants (5). It is associated with several congenital syndromes, including sporadic aniridia, isolated hemihypertrophy, the Denys-Drash syndrome (nephropathy, renal failure, male pseudohermaphroditism, and Wilms’ tumor), genital anomalies, Beckwith-Wiedemann syndrome (visceromegaly, macroglossia, omphalocele, and hyperinsulinemic hypoglycemia in infancy), and the WAGR complex (Wilms’ tumor with aniridia, genitourinary malformations, and mental retardation), which suggested a genetic predisposition to this tumor (6,7). Wilms’ tumor is
also reported in individuals with Simpson-Golabi-Behmel syndrome, another overgrowth syndrome similar to Beckwith-Wiedemann in many respects (8). These congenital disorders have now been linked to abnormalities at specific genetic loci implicated in Wilms’ tumorigenesis.
also reported in individuals with Simpson-Golabi-Behmel syndrome, another overgrowth syndrome similar to Beckwith-Wiedemann in many respects (8). These congenital disorders have now been linked to abnormalities at specific genetic loci implicated in Wilms’ tumorigenesis.
GENETIC ORIGINS
The identification of a chromosomal deletion of band p13 of the eleventh chromosome in children with the WAGR syndrome led to a search at this site for a Wilms’ tumor-associated gene. Subsequent molecular studies of children with Wilms’ tumor have demonstrated, in some cases, a deletion at this site (9). This deletion has subsequently been shown to include the aniridia gene and a putative Wilms’ tumor suppressor gene (WT1). The protein product of this gene has subsequently been characterized and is a developmentally regulated transcriptional factor of the zinc finger family that regulates the expression of other genes, including growth-inducing genes such as those encoding early growth response, insulinlike growth factor 2 (IGF2), and platelet-derived growth factor A chain (10,11). Suppression of these growth-associated genes may explain the tumor suppressor role of WT1. Recently, the WT1 gene product has been found to physically bind to the p53 protein (12). The WAGR syndrome has been correlated with constitutional deletions of band q13 and virtually all patients with Denys-Drash syndrome carry point mutations in WT1 in the germline (9,13). These result in a dominant negative oncogene and more severe somatic abnormalities than in the WAGR syndrome.
Abnormalities at the 11p15 locus have been associated with Beckwith-Wiedemann patients, although the locus for a Wilms’ tumor gene has not been defined, nor is it known whether the Beckwith-Wiedemann locus and WT2 are the same or contiguous loci. Some children with Beckwith-Wiedemann syndrome have overexpression of a gene normally expressed by only one of the parental alleles. In some cases, a constitutional duplication of the paternal 11p15 chromosomal fragment has been identified (trisomy at 11p15) (14). In other cases, both copies are from the father with none from the mother (uniparental isodisomy) (15,16). These findings led to speculation that the Beckwith-Wiedemann gene is expressed only by the paternal allele, and these genetic abnormalities that lead to the presence of two paternal alleles would double the expression of this gene and may result in the overgrowth. Mutations in the tumor-associated genes WT1 and WT2 do not appear to have any prognostic significance for children with Wilms’ tumor.
Two WT2 candidate genes are the IGF2 gene and the H19 gene. The IGF2 gene is present at the 11p15 locus. It is an embryonal growth-inducing gene with expression restricted to the paternal allele (17). However, there is no direct evidence to link the IGF2 gene to the causation of the Beckwith-Wiedemann syndrome (18). It is of note that children whose manifestations of the Beckwith-Wiedemann syndrome include hemihypertrophy appear to have a greater risk for the occurrence of malignancy than those who do not. In a series reported by Wiedemann, cancer was reported in 7.5% of all children with the syndrome, but in more than 40% of children with both the syndrome and hemihypertrophy (19). Loss of expression of H19, a tumor suppressor gene, has also been reported in Wilms’ tumors (20). The H19 gene is expressed from the maternal allele. With loss of heterozygosity, the cell may lose the maternal (active) copy and hence its tumor suppressor function.
Familial cases of Wilms’ tumor account for only 1% to 2% of patients with Wilms’ tumor and have not been associated with the previous syndromes. Analysis of two families revealed a link with chromosome band 17q12–21, and the putative tumor gene at this locus has been named FWT1 (21,22). More recent studies have demonstrated in five kindreds an inherited Wilms’ tumor predisposition gene at 19q13.3–q13.4 called FWT2 (23). In addition, loss of heterozygosity was see at 19q in tumors from individuals from two families whose predisposition is not due to this 19q locus, suggesting that alterations at two distinct loci are critical rate-limiting steps in the etiology of these familial Wilms’ tumors involving both germline predisposing mutations and somatic alteration at a second focus. The Simpson-Golabi-Behmel syndrome is a sex-linked syndrome linked to Xq25–27 and the protein product Glypican 3 may interact with IGF2 receptor (24,25).
More recent studies have suggested that loss of heterozygosity on chromosome 16q in Wilms’ tumors (observed in 15% to 20% of cases) was associated with a 3.3 times greater incidence of relapse and a 12 times greater incidence of mortality, as compared with children without these chromosomal changes (26). A similar trend was seen for children with loss of heterozygosity for 1p, which occurs in approximately 10% of Wilms’ tumors, but these trends were not statistically significant. One of the primary goals of the fifth NWTS study was to assess whether several identified chromosomal abnormalities were of prognostic significance in Wilms’ tumor and might provide some guidance for future therapeutic recommendations.
Routine radiographic screening for children with syndromes associated with Wilms’ tumor has been recommended. Ultrasonograms are generally obtained every 3 months until the children are 5 years of age. No prospective studies, however, have been performed to evaluate the cost effectiveness or efficacy of following this recommendation (27,28). Retrospective reviews of routine ultrasonographic screening report conflicting results with regard to clinical benefit from such screening, as judged by the tumor stage distribution at presentation or the outcome of the children (29,30).
PATHOLOGIC PRECURSORS: NEPHROGENIC RESTS, NEPHROBLASTOMATOSIS, AND MULTICYSTIC DYSPLASTIC KIDNEYS
The presence of nephrogenic rests (NR; persistent metanephric tissue in the kidney after the thirtysixth week of gestation) has been associated with the occurrence of Wilms’ tumor. They may occur in a perilobular (PLNR) or intralobular (ILNR) location and may be single or multiple (Fig. 35-1). In children with aniridia or Denys-Drash syndrome, the lesions are primarily ILNR, whereas children with hemihypertrophy or the Beckwith-Wiedemann syndrome have predominantly PLNR (31). The presence of multiple or diffuse nephrogenic rests is termed nephroblastomatosis.
An autopsy series of infants under 3 months of age defined the frequency of nephrogenic rests. Nine of 1,035 infants (0.87%) had PLNRs, and ILNRs occurred in only 2 of 2,000 cases (0.1%) (32). Most nephrogenic rests when identified are sclerosing and in an apparently indolent or involutional phase. The vast majority will spontaneously resolve without producing a tumor as the incidence of nephrogenic rests is about 100 times greater than that of Wilms’ tumor (1/10,000 infants).
In addition to their anatomic site, nephrogenic rests are also classified histologically as incipient or dormant nephrogenic rests, regressing or sclerosing nephrogenic rests, and hyperplastic nephrogenic rests (Fig. 35-2) (33). Incipient or dormant rests are composed predominantly of blastemal or primitive epithelial cells resembling those seen in embryonic kidney and Wilms’ tumor, but are microscopic with sharp margins from adjacent renal parenchyma. The term incipient is used in infants and young children, whereas dormant is used in older children. The regressing or sclerosing rests demonstrate maturation of the cellular elements and progress to obsolescent rests, which are composed primarily of hyalinized stromal elements. Nephrogenic rests of the hyperplastic variant are often difficult to distinguish from small Wilms’ tumors. They
demonstrate diffuse or synchronous proliferation of components throughout the rest. This uniform growth leads to preservation of the original shape of the rest in contrast with neoplastic proliferation of a single cell that produces a more spherical expanding nodule within the rest. Incisional or needle biopsies of these lesions cannot be reliably distinguished from a Wilms’ tumor, and preservation of the shape of the original rest is the most obvious clue that one is dealing with a hyperplastic, rather than a neoplastic, change (33). Histologically, most hyperplastic nodules lack a pseudocapsule at their periphery, whereas most Wilms’ tumors will have one. Hence, excisional biopsies that contain the margin are required to adequately differentiate between hyperplastic nephrogenic rests and Wilms’ tumor.
demonstrate diffuse or synchronous proliferation of components throughout the rest. This uniform growth leads to preservation of the original shape of the rest in contrast with neoplastic proliferation of a single cell that produces a more spherical expanding nodule within the rest. Incisional or needle biopsies of these lesions cannot be reliably distinguished from a Wilms’ tumor, and preservation of the shape of the original rest is the most obvious clue that one is dealing with a hyperplastic, rather than a neoplastic, change (33). Histologically, most hyperplastic nodules lack a pseudocapsule at their periphery, whereas most Wilms’ tumors will have one. Hence, excisional biopsies that contain the margin are required to adequately differentiate between hyperplastic nephrogenic rests and Wilms’ tumor.
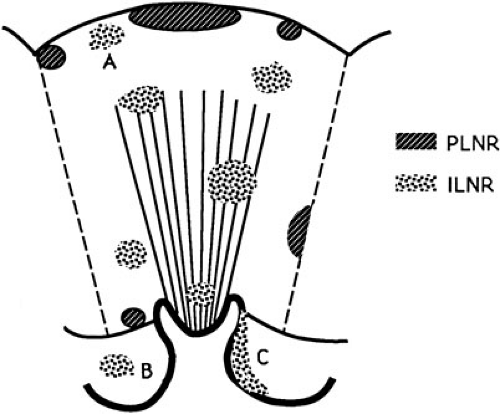 FIGURE 35-1. Diagrammatic depiction of nephrogenic rests and their classification. Heavy arrows indicate tumor induction. (Adapted from Beckwith JB. Precursor lesions of Wilms’ tumor: clinical and biological implications. Med Pediatr Oncol 1993;21:158–168, with permission.) |
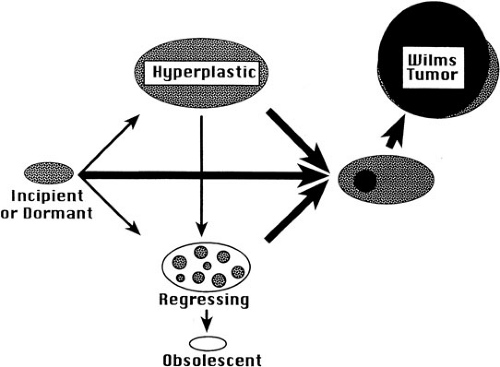 FIGURE 35-2. Diagram of a renal lobe and adjacent calyx and intervening sinus with potential sites of distribution of perilobar (PLNR) and intralobar (ILNR) nephrogenic rests including the superficial cortex (A), renal sinus (B), and pelvicalyceal wall (C). (Adapted from Beckwith JB, Kiviat NB, Bonadio JF. Nephrogenic rests, nephroblastomatosis, and the pathogenesis of Wilms’ tumor. Pediatr Pathol 1990;10:1–36, with permission.) |
Despite their relatively rare occurrence, nephrogenic rests are frequently found in association with Wilms’ tumors. In a review of unilateral Wilms’ tumors in the National Wilms’ Tumor Study-4 (NWTS-4), 41% of unilateral tumors were associated with nephrogenic rests (31). In children with synchronous bilateral Wilms’ tumor the incidence of nephrogenic rests was 99%. These were primarily PLNRs—possibly due to the fact that these lesions are much more prevalent than the ILNRs. Similarly, an increased incidence of nephrogenic rests is seen in children with the syndromes associated with Wilms’ tumor, which were discussed previously (Table 35-1) (33).
Gylys-Morin and colleagues demonstrated that magnetic resonance imaging (MRI) scans can be helpful in following children with nephroblastomatosis (34). Alterations in imaging characteristics of the lesions suggest a transition from nephrogenic rests to Wilms’ tumor, as does growth of isolated lesions.
Diffuse hyperplastic perilobar nephroblastomatosis (DHPLN) is a distinct entity that must be distinguished clinically from Wilms’ tumor. Infants with this entity may present with unilateral or bilateral large flank masses (Fig. 35-3). A characteristic finding is massively enlarged kidneys that maintain their normal configuration and lack necrosis. As with the isolated nephrogenic rests, proliferation of the thin rind of nephrogenic rests on the periphery of the tumor will preserve the normal configuration of the kidney, but with marked enlargement of its size. This is in contrast with Wilms’ tumor where the normal renal configuration is generally distorted. Nephrectomy is not required in these cases. Chemotherapy has been employed to control the proliferative element of the nephrogenic rests. Its use may accelerate the resolution in size of the masses and decrease the respiratory compromise they may produce. It has not, however, been established that treatment with chemotherapy will decrease the risk of malignancy occurring in these lesions. A more recent review of 65 cases of DHPLN revealed the subsequent development of Wilms’ tumor in 19 children from 2 to 79 months following presentation (median, 30 months). The histology of these tumors was favorable in 12, diffuse anaplasia in 5, and focal anaplasia in 2—a surprisingly high incidence of anaplasia (35). It has been suggested that Wilms’ tumors have an increased occurrence in multicystic dysplastic kidneys. The frequency of nephrogenic rests in multicystic dysplastic kidneys is estimated to be 4%, approximately five times the prevalence in a random autopsy population of infants under 3 months of age (36). If one were to estimate the frequency of Wilms’ tumor in these kidneys, the standard risk of 1 in 10,000 infants might be said to be increased to 1 in
2,000. Review of the NWTS pathology files identified only 3 cases of dysplastic kidneys in more than 7,000 children with Wilms’ tumor over a 26-year interval and only 1 case in more than 1,500 referral cases sent to Dr. Beckwith from around the world. Although it is impossible to estimate the number of children at risk of developing Wilms’ tumors in remaining dysplastic kidneys, it must be concluded that the risk of development of Wilms’ tumor in kidneys with multicystic dysplasia or congenital obstruction must be extremely low and does not justify nephrectomy to avoid the development of tumors.
2,000. Review of the NWTS pathology files identified only 3 cases of dysplastic kidneys in more than 7,000 children with Wilms’ tumor over a 26-year interval and only 1 case in more than 1,500 referral cases sent to Dr. Beckwith from around the world. Although it is impossible to estimate the number of children at risk of developing Wilms’ tumors in remaining dysplastic kidneys, it must be concluded that the risk of development of Wilms’ tumor in kidneys with multicystic dysplasia or congenital obstruction must be extremely low and does not justify nephrectomy to avoid the development of tumors.
TABLE 35-1 Association of Nephrogenic Rests with Wilms’ Tumor and Associated Syndromes. | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
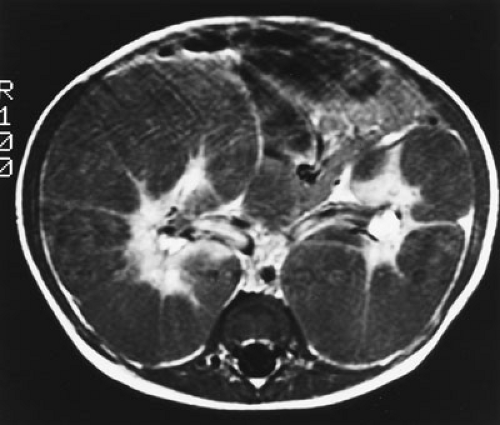 FIGURE 35-3. Magnetic resonance imaging scan of a 10-month-old infant who presented with large bilateral flank masses. It demonstrates a picture characteristic of diffuse hyperplastic perilobar nephroblastomatosis (DHPLN) with extensive involvement of the entire cortex of the kidney with no evidence of necrosis and general preservation of the shape of the kidney. |
PATHOLOGY OF RENAL TUMORS
The collection of large numbers of renal tumor specimens by the cooperative group trials has allowed the development of accurate pathologic classifications in a much shorter period of time than would have ever been feasible without these trials. Early reports of Wilms’ tumors and the initial cooperative group trials included essentially all renal sarcomas under this rubric, but time and experience has now allowed identification of several subgroups of tumors that are at particularly high risk of recurrence and adverse outcome in contrast with the vast majority of cases that respond to standard therapy (37,38). In NWTS-1, anaplastic and sarcomatous variants comprised only 11.5% of the tumors, yet they accounted for 51.9% of deaths due to tumor. These unfavorable histologies proved to be the most important factor in patient outcome in NWTS-1 and that finding continues through the current studies. Wilms’ tumors are currently divided into those with “favorable” histology and those with “unfavorable” histology (Table 35-2). The latter group includes tumors with focal or diffuse anaplasia (39,40). Clear cell sarcoma of the kidney and malignant rhabdoid tumors of the kidney were initially grouped with the unfavorable histology Wilms’ tumors, but are now considered as distinct entities based on their pathologic appearance and response to quite different therapies (41,42,43,44).
TABLE 35-2 Pathologic Classification of Renal Tumors. | |
|---|---|
|
TABLE 35-3 Staging System Used by the National Wilms’ Tumor Study Group. | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The staging system used by the NWTS group is a pretreatment surgical staging system (Table 35-3). It must be carefully distinguished when comparing treatment results with children treated on the SIOP protocols where the staging information is obtained after preliminary treatment of the tumors (Table 35-4). The intensity of adjuvant treatment is determined by such factors as regional lymph node involvement and penetration of the renal capsule by tumor, which cannot be accurately determined by radiographic studies. The staging criteria have been adjusted during the course of the NWTS group studies as the prognostic significance of criteria were established (45).
TABLE 35-4 Staging System Used by the Societe Internationale d’Oncologie Pediatrique, Based on Findings after Preoperative Therapy. | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
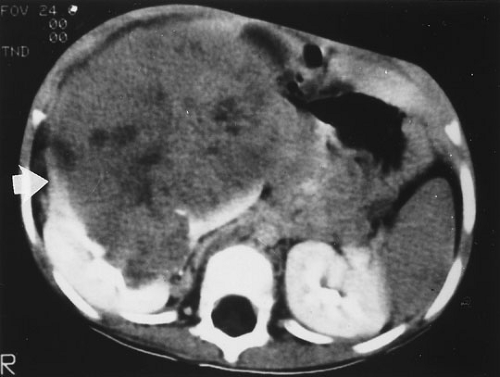 FIGURE 35-4. A computed tomography scan of a 3.5-year-old boy who presented with a large abdominal mass demonstrating the characteristic findings of a Wilms’ tumor. The tumor mass can be seen protruding from the right kidney with a margin of renal parenchyma along the periphery (arrow). |
Wilms’ tumor is characterized as a triphasic embryonal neoplasm with blastemal, stromal, and epithelial components (Fig. 35-4) (38). Each component can express several patterns of differentiation that lead to several histologic subgroups of Wilms’ tumors. One particular subtype, the fetal rhabdomyomatous nephroblastoma, has been associated with poor response to chemotherapy, but a generally good prognosis (46). Large, pleomorphic and hyperchromatic nuclei with abnormal multipolar mitotic figures characterize the anaplastic tumors. Anaplasia can occur in the epithelial, stromal, or blastemal populations, or any combination of these three. This pattern occurs primarily in children older than 2 years of age. In NWTS-1, 66.7% of the patients relapsed and 58.3% succumbed to their tumor (37). Even in this early report, the distinct implications of the “diffuse” versus the “focal” pattern was appreciated with a higher frequency of relapse and death in the “diffuse” subgroup. This was confirmed in review of the NWTS-2 and NWTS-3 studies. Although children with stage I anaplastic tumors generally did well, children with stage II to IV tumors did poorly. The severity of dysplasia was not a predictive factor, however, anaplasia in extrarenal tumor sites and a predominantly blastemal tumor pattern were both adverse prognostic factors (47).
Approximately 1% of children presenting with a unilateral tumor will develop contralateral disease. Fifty-eight of 4,669 children registered in the first four NWTS group studies developed metachronous disease (48). Analysis of this cohort by a matched case control study demonstrated that the children with nephrogenic rests had a significantly increased risk of metachronous disease, particularly those with PLNRs. This finding was especially true for young children where a Wilms’ tumor occurred in 20 of 206 children under 12 months old in comparison to 0 of 304 children older than 12 months old. These young infants younger than 12 months of age with Wilms’ tumor who also have nephrogenic rests require frequent surveillance for several years for the development of contralateral disease.
Clear cell sarcoma of the kidney is a highly malignant tumor with an unusual proclivity to bony metastasis. It generally presents as a large unifocal and unilateral tumor with homogeneous mucoid, tan or gray-tan cut surface, often with foci of necrosis or prominent cyst formation (38,49). This tumor invades surrounding renal parenchyma rather than compressing it into a pseudocapsule as occurs with a Wilms’ tumor. Its classic appearance is that of a deceptively bland tumor with uniform oval nuclei with a delicate chromatin pattern and a prominent nuclear membrane and sparse poorly stained vacuolated “water-clear” cytoplasm with indistinct cell membranes. The cells often appear in cords or nests divided by an arborizing network of vessels and supporting spindle cell septa, although nine major histologic patterns have been identified (49). The cell of origin for this tumor is not known.
Malignant rhabdoid tumors of the kidney occur in young infants at a median age of 11 months (44). Eighty-five percent of the cases occur within the first 2 years of
life. A characteristic involvement of the perihilar renal parenchyma is seen. Histologically, they are characterized by the presence of monomorphous, discohesive, rounded to polygonal cells with acidophilic cytoplasm and eccentric nuclei containing prominent large “owl eye” nucleoli reminiscent of skeletal muscle, but lacking its cytoplasmic striations, ultrastructural features, and immunochemical markers (38). A large PAS-positive hyaline, cytoplasmic inclusion occurs in a variable population of tumor cells and is seen as a hallmark of this tumor (42). Ultrastructural examination reveals parallel cytoplasmic filamentous inclusions packed in concentric whorled arrays, a distinctive feature of this tumor that suggests a neuroectodermal origin. The tumor tends to infiltrate surrounding renal parenchyma rather than compress it. These tumors are notable for the occurrence of second primary neuroglial tumors in the midline of the brain resembling medulloblastoma (50). A consistent deletion of 22q11–12 has been described in both renal and extrarenal rhabdoid tumors (51,52).
life. A characteristic involvement of the perihilar renal parenchyma is seen. Histologically, they are characterized by the presence of monomorphous, discohesive, rounded to polygonal cells with acidophilic cytoplasm and eccentric nuclei containing prominent large “owl eye” nucleoli reminiscent of skeletal muscle, but lacking its cytoplasmic striations, ultrastructural features, and immunochemical markers (38). A large PAS-positive hyaline, cytoplasmic inclusion occurs in a variable population of tumor cells and is seen as a hallmark of this tumor (42). Ultrastructural examination reveals parallel cytoplasmic filamentous inclusions packed in concentric whorled arrays, a distinctive feature of this tumor that suggests a neuroectodermal origin. The tumor tends to infiltrate surrounding renal parenchyma rather than compress it. These tumors are notable for the occurrence of second primary neuroglial tumors in the midline of the brain resembling medulloblastoma (50). A consistent deletion of 22q11–12 has been described in both renal and extrarenal rhabdoid tumors (51,52).
The occurrence of primitive neuroectodermal tumor (PNET) of the kidney is well documented (53). It is clearly distinct from Wilms’ tumor and the other variants previously discussed, and demonstrates spread to lymph nodes, lung, bone, liver, and bone marrow, as is seen when PNET occurs in other anatomic locations (54). Its treatment including resection, chemotherapy, and radiation must follow that of PNET and not that of other renal tumors.
TREATMENT
As a result of the work by Sidney Farber that demonstrated the role of adjuvant therapy in Wilms’ tumor, all children treated on early protocols received chemotherapy. Only since the mid-1990s, has adjuvant therapy been avoided in a small proportion of children identified to have an extremely low risk of local recurrence and metastasis. Optimal chemotherapy regimens have been established in a series of well-designed randomized studies primarily performed by the NWTS group in the United States and Canada and SIOP in Europe. Despite advances in chemotherapy, surgery continues to play a critical role in the treatment of Wilms’ tumor. Accurate staging and safe and complete resection of the tumor are key elements in achieving cure. Local control is rarely achieved by chemotherapy and radiotherapy alone.
Chemotherapy
Wilms’ tumor was the first malignant pediatric solid tumor found to be responsive to dactinomycin (3). Many additional agents have been subsequently identified: vincristine, doxorubicin, cyclophosphamide, ifosphamide, and etoposide.
Children with stage I tumors were treated on the third NWTS group protocol (NWTS-3) with an 11-week regimen composed of vincristine and dactinomycin without abdominal radiation, based on the results of the initial two NWTS studies. The 4-year relapse-free survival (RFS) and overall survival were 89.0% and 95.6%, respectively (55). The other three stages were treated on protocols that involved randomization of two or four arms. This study supported the addition of doxorubicin to the treatment of children with stage III tumors, but did not demonstrate any benefit to the addition of doxorubicin or radiotherapy for children with stage II tumors or benefit to the addition of cyclophosphamide to the treatment of children with stage IV tumors.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


