Principles of Oncology
Lesley Simpson
Kristin Baird
David N. Korones
Cindy L. Schwartz
Sidney Kimmel Cancer Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, Maryland 21287.
Pediatric M&D Hematology/Oncology, University of Rochester, Rochester, New York 14627.
Childhood malignancy is a relatively rare event, affecting 1 child in 500 by 15 years of age (1), and 1 in 300 to 330 by age 19. Although cancer remains the second leading cause of death, after accidents, for children 14 years of age and younger, the past quarter of a century has seen remarkable advances in treatment. Cure rates have increased from 28% in 1963 to more than 75% in the 1990s. As a result, 1 in 400 young adults are survivors of cancer diagnosed before age 20.
A multidisciplinary approach to the care of the oncology patient has contributed to many advances. Wilms tumor has been described as a prism through which our knowledge of oncologic principles can be viewed (2). Pediatric oncologists, surgeons, pathologists and radiotherapists tackled the problem of Wilms tumor together and set the stage for our modern era by forming a cooperative group, the National Wilms’ Tumor Study (NWTS), to investigate treatment approaches. Survival rates soared from 20% with surgery alone to 50% with the addition of radiotherapy and then to 90% with the chemotherapeutic advances of the NWTS (3). As survival became commonplace, oncologists became aware of long-term problems, such as scoliosis after radiotherapy and cardiotoxicity after treatment with doxorubicin. Large cohorts of patients treated by the NWTS groups allowed for assessment of the relative benefits of components of multimodality therapy. More toxic therapies could be eliminated in appropriate cohorts. Likewise, acute lymphoblastic leukemia, the most common cancer in children, has become curable for 80% of children as a result of cooperative group trials (4). With this success, trials can now focus on improving therapy for high-risk disease, while decreasing toxicity for low-risk disease.
Much of the early success was achieved with only limited understanding of the pathogenesis of the uncontrolled proliferation of cells that characterizes malignancy. A rapid and ongoing expansion of our understanding of cancer is likely to improve not only rates of cure, but also the specificity with which we can halt the growth of malignant cells. Although it has been possible to decrease toxicity by dose modification in some tumors, in other tumors, improvements in survival have been achieved only by dose intensification and increased toxicity. It becomes increasingly clear that treatments must become more specifically designed to kill the abnormal cells, while preserving the normal cells. This specificity can be achieved only with better understanding of the biologic difference between neoplastic and normal cells.
This chapter explores the genetic and environmental basis for cancer, as well as principles of tumor cell growth and regulation, the roles of radiotherapy and chemotherapy in treatment, and the causes of treatment failure. Because cytotoxic therapies will remain a major part of our armamentarium for years to come, the long-term effects on normal tissues will continue to plague these patients. Late effects of cancer therapy are discussed at the end of this chapter.
THE BIOLOGY AND GENETICS OF CANCER
“Why did my child get cancer?” The question that every parent asks drives oncology research worldwide. This section explores the genetic and environmental basis of cancer development, as well as the biology of tumor growth—the roles of mutations, differentiation, and metastasis—the growth cycle, and the growth kinetics of tumor masses.
Genetic Causes
Over the past several decades, it has become clear that cancer is the result of DNA mutations in tumor cells (5). Many cancer-causing genes have now been cloned and sequenced. Our increased understanding of these genes is beginning to allow for drug design and gene targeted therapy that will specifically block cancer cell growth.
Growth Cycle
The process by which one cell becomes two is highly coordinated and complex, governed by multiple cell cycle-related proteins, allowing for checks and counterchecks to ensure faithful transmission of genetic information. The gain or loss of function of any of these proteins constitutes the molecular basis of oncogenesis. A brief discussion of normal growth cycle control is helpful in understanding how cancer cells deviate from the normal process (6).
The cell growth cycle is arbitrarily divided into five phases, each of which is characterized by a distinct cellular activity related to cell proliferation (Fig. 32-1). After completion of mitosis, cells enter either G0 or G1 (the G stands for gap). Those that enter G0 are no longer a part of the growth cycle. Instead, they are fully differentiated cells, such as hepatocytes or neurons that, under normal circumstances, remain permanently in G0 carrying out the specific functions of their differentiated phenotypes. Other cells rest in G0 but can be stimulated to reenter the growth cycle. For example, mature lymphocytes are in G0, but with the appropriate stimulus, mature lymphocytes move into the growth cycle and proliferate. Tumor cells, too, can move in and out of G0.
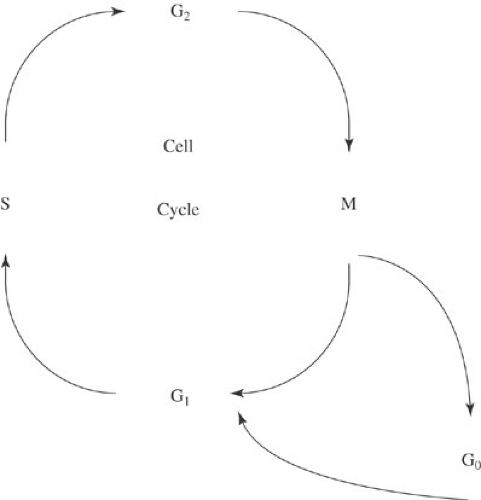 FIGURE 32-1. The cell cycle. S, DNA synthesis; M, mitosis; G0, quiescent phase; G1, synthesis of enzymes and factors needed for DNA synthesis; G2, synthesis of enzymes and factors needed for mitosis. |
Cells can enter G1 either directly from mitosis or after resting in G0. G1 is the gap between completion of mitosis and initiation of DNA synthesis. Enzymes and other factors necessary for DNA synthesis are produced during this phase. The amount of time a tumor cell spends in G1 is a critical determinant of the doubling time of the tumor. After G1, cells enter S phase, the period of DNA synthesis during which the entire genome is precisely replicated. Because a relatively high proportion of cancer cells (compared with normal cells) are in S phase, many chemotherapeutic agents are designed to be S-phase specific—that is, they inhibit DNA synthesis. Cells then enter G2, the period between DNA synthesis and mitosis. Enzymes and other factors necessary for mitosis are synthesized during G2. G2 is followed by mitosis, the process by which the two identical sets of chromosomes produced in S phase are divided into each of two daughter cells. After completion of mitosis, the cycle begins anew, becoming quiescent (G0) or reentering the replication cycle (G1).
Mutations: Oncogenes and Tumor Suppressor Genes
The behavior of tumor cells is not static: They can become quiescent or differentiate; they can die or grow more rapidly; and they can acquire features that predispose them to metastasize. These changes in behavior are usually caused by genetic mutations. In vitro studies of proliferating cells show a mutation rate of about one mutation per gene per 106 normal cells and about one mutation per gene per 105 cancer cells (7). Thus, there appears to be about a 10-fold increased mutations rate in malignant cells. A majority of cancer genes control, directly or indirectly, cell cycle progression—the oncoproteins encoded by these genes disrupt DNA replication error repair, apoptosis, checkpoints, and the G1–S transition. In general, two classes of cancer genes have been identified: overexpressed protooncogenes that promote tumorigenesis, and underexpressed tumor suppressor genes that lose the ability to regulate cell growth (8).
Protooncogenes are often formed by chromosomal translocations that bring together regions of different chromosomes to produce fused transcription factors with altered function. Examples include the Mll fusion genes in leukemias, t(4,11) and t(9,11); the Ews fusion gene t(11,22) in childhood sarcomas; and the Bcr-abl fusion gene t(9,22) in chronic myelogenous leukemia (CML) and acute lymphoblastic leukemia (ALL). The products of tumor suppressor genes normally provide negative controls of the cell cycle. Loss of function through mutation or deletion eliminates this cell cycle control and allows for malignant transformation. Examples include RB gene, mutated in retinoblastoma, osteosarcoma, and other soft tissue
sarcomas; p53, expressed in all human cells and inactivated in colon, lung, breast, stomach, liver, ovary, and prostate cancers; and finally, WT1 is mutated in some Wilms’ tumors and leukemias.
sarcomas; p53, expressed in all human cells and inactivated in colon, lung, breast, stomach, liver, ovary, and prostate cancers; and finally, WT1 is mutated in some Wilms’ tumors and leukemias.
Kinetics of Tumor Growth
Much of what we know about the kinetics of tumor growth is derived from mathematic and animal models developed several decades ago. Skipper (9). established two important principles of tumor growth, known as Skipper’s laws, based on observations of the growth of a murine leukemia cell line in mice. The first of these principles is that the doubling time of a given cancer cell or mass of cancer cells is constant. Thus, if it takes 24 hours for one cancer cell to become two, it will take the same amount of time for 1,000 cells to become 2,000. The second principle is that the percentage of tumor cells killed by a drug is constant, regardless of the number of tumor cells present. For example, if a certain drug kills 90% of a mass of cells, that drug would reduce a tumor burden of 100 cells to 10, or 1,000,000 cells to 100,000.
Although Skipper’s laws still apply to growing tumors, they have been substantially modified. The first modification came in 1960, when Mendelsohn (10) introduced the concept of growth fraction. The growth fraction is the proportion of a mass of tumor cells that are in the growth cycle (as opposed to G0). Analysis of tumor cells by flow cytometry or tritiated thymidine labeling has enabled investigators to estimate growth fractions. The growth fraction ranges from less than 10% in slow-growing tumors, such as low-grade astrocytoma, (11) to 70% to 90% in more aggressive tumors, such as Burkitt’s lymphoma (12). Thus, Skipper’s laws of cell growth and cell kill do not apply to an entire tumor as originally postulated, but only to the fraction of tumor cells in the growth cycle.
Metastasis
Metastasis is a major cause of treatment failure and death in children with cancer. In fact, local control of primary tumors with radiation and surgery is so effective for many of the pediatric malignancies that chemotherapy is largely directed toward control of metastases or micrometastases. The process of metastasis is an intricate, complex, and incompletely understood phenomenon. Formation of metastases is similar among different tumor types (13). First, progressive growth of neoplastic cells must take place. Second, extensive vascularization must ensue to provide nutrition to this expanding mass. Once the tumor has grown beyond 1 mm in diameter, it relies on angiogenesis to sustain its growth. Third, local invasion into the normal tissue takes place via direct penetration of thin walled venules. Then tumor cells must detach and emobolize through the circulation. They must then adhere to capillary endothelial cells or basement membranes in distant organs. Finally, extravasion and proliferation with the distant organ completes the metastatic process.
Exciting advances in our understanding of these pathologic events of metastasis may lead to therapy specifically designed to prevent it. For example, antiangiogenesis agents have been designed to block the vascularization required for initial tumor growth. Other agents attempt to disrupt adhesion molecules on the surface of cancer cells or their receptors on basement membranes of distant organs.
PRINCIPLES OF RADIOTHERAPY
DNA damage is responsible for the cytotoxic effects of radiotherapy. Radiation produces a direct effect on the DNA via double-strand breaks. However, the most prominent cytotoxic effect is indirect. The ionization of water in cells produces reactive free radicals, such as hydroxyl and oxygen radicals, peroxide, and hydrated electrons. These free radicals then disrupt the DNA sugar backbone and its bases. Cells are most sensitive to radiotherapy in the early S phase (DNA synthesis) of the cell cycle and in mitosis when the cells’ inability to replicate prevents progression to the next phase. Rapidly dividing cells die rapidly and thus are more radiosensitive. The cells of the oral and digestive mucosa are rapidly dividing and show damage within a few weeks of radiation. The lympholytic effect of radiation can cause rapid regression of lymphomas. More slowly dividing cells are more tolerant to radiation effect. Slowly dividing cells, like brain cells, may not manifest radiation damage for months or years after therapy. Similarly, slow-growing tumors, like some sarcomas, may be less responsive to radiation.
The efficacy of radiotherapy is also related to the presence of oxygen in the target tissue, allowing production of radiation-induced free radicals. Tissue hypoxia, common in tumors, results in substantially reduced cell killing. Tumor cells that are hypoxic at the time of radiation require a higher dose to achieve the same level of cytotoxicity, or they may be radiation resistant (14).
The variables that influence therapeutic response include the total dose, the number of fractions, and the duration of therapy. Radiation is traditionally administered in small fractions of the total dose to allow for protracted treatments. It is believed that multiple small fractions allow for repair of sublethal damage, particularly in normal tissues that repair sublethal damage better than do tumors. Fractionation of the radiation also results in prolongation of treatment time, allowing hypoxic areas of tumor tissue to become reoxygenated and more radiosensitive.
Radiation therapy can be broadly divided into external beam and brachytherapy (15). Photon beams use electrons accelerated along a wavelength to produce high-energy X-rays. The photon energy is absorbed in tissue, and beam
penetration is proportional to energy level. Electron beam energy is absorbed more superficially than from photon beams, allowing for uniform dose over the effective range. Proton, or heavy particle, irradiation allows discrete, defined high-dose localization. Brachytherapy describes direct application of radionucleotide seeds (192Ir and 125I) to surgically accessible sites. It allows for confined, continuous doses to tumors with relative sparing of adjacent tissue.
penetration is proportional to energy level. Electron beam energy is absorbed more superficially than from photon beams, allowing for uniform dose over the effective range. Proton, or heavy particle, irradiation allows discrete, defined high-dose localization. Brachytherapy describes direct application of radionucleotide seeds (192Ir and 125I) to surgically accessible sites. It allows for confined, continuous doses to tumors with relative sparing of adjacent tissue.
Various radioisotopes are being investigated for systemic delivery of targeted radiotherapy. Samarium-153, for example, has a high affinity for skeletal tissue and can concentrate in bone with high metabolic activity. High doses can be delivered to regions with enhanced osteoblastic activity (i.e., bony metastases) over a short period of time, with little residual activity in the bone marrow.
The use of radiotherapy requires knowledge of tumor sensitivity and tolerance to radiation of the normal adjacent tissues. Young children are particularly sensitive to long-term effects of radiotherapy, in part because of their increased growth potential. Although radiotherapy can be used as the sole modality of treatment for tumors such as Hodgkin’s disease and brain tumors, the use of a single modality for the treatment of malignancy is becoming less common. Combined-modality therapy often offers the possibility of limiting total radiation dose while improving cure rates.
PRINCIPLES OF CHEMOTHERAPY
The ideal drug would be one that is targeted to block a tumor-specific physiologic process and has little effect on other organs or physiologic processes. Unfortunately, despite significant progress in the understanding of the molecular and genetic basis of the development of cancer, the evolution of targeted cancer therapy has been slow and newer, more effective agents have been difficult to identify. We have had to rely primarily on cytotoxic chemotherapy and the basic principles of tumor growth and development for the treatment of cancer. Many of the drugs designed to kill cancer cells also kill normal cells because cancer cells are not sufficiently different from normal cells. Often, however, enough differences exist between the two cell types to provide a therapeutic window for preferential tumor cell killing, albeit a narrow one. The principles of cytotoxic chemotherapy are based on the relatively rapid growth rate of tumor cells, the gompertzian growth kinetics of tumor masses, and the propensity of tumor cells to undergo mutations and become more resistant to subsequent treatment. These principles provide the rationale for treating cancer promptly with high doses of drugs, with combination chemotherapy, and typically in a cyclic pattern. More recently, researchers and clinicians have focused on two new areas of potential cancer therapeutics and incorporated these into existing therapeutic regimens. One area, immunotherapy, is based on the principle that the host immune system can be stimulated and modulated to help identify and destroy tumor cells. The second area focuses on specific molecular mechanisms intrinsic to the tumor in order to devise targeted therapies that directly attack the tumors while sparing normal tissue. The ultimate goal is to develop more effective therapies with far less toxicity than is typically associated with cancer chemotherapy.
Drug Development
The ultimate goals of cancer chemotherapy can be stated simply: (1) to cure cancer and (2) to cause little or no side effects. High throughput drug screening has been the predominant mechanism of cancer drug development over the past several years. This method incorporates exposing multiple cancer cell lines to compounds with suspected or known cytotoxic effects. Those drugs with the most profound apoptotic and growth inhibitory effects are then selected to go onto preclinical and clinical trials. The initial step is a phase I clinical trial. During phase I testing, the maximum tolerable dose is established and the agent’s toxicity profile is identified. In phase II clinical trials, previously established maximal tolerated doses are used to establish the efficacy of an agent in particular malignancies. Once dosing and efficacy are established, phase III studies are designed to compare the new agent or regimen to an established regimen. This may be a direct comparison of two agents or an evaluation of the impact of integrating a new agent into an established regimen.
TRADITIONAL CYTOXIC CHEMOTHERAPY
Cell Proliferation
Virtually all standard antineoplastic agents interfere with cell proliferation. At the level of DNA synthesis, antimetabolites, such as methotrexate and 5-fluorouracil, inhibit synthesis of purines and pyrimidines. Alkylating agents, such as cyclophosphamide, mechlorethamine (nitrogen mustard), and carboplatin, covalently bind to DNA and prevent replication and transcription. L-Asparaginase inhibits protein synthesis, and vincristine and paclitaxel (Taxol) interfere with mitotic spindle formation and breakdown. Most of these events occur during G1, S, G2, or M phase of the growth cycle. Thus, cells in G0 are impervious to many of the standard therapies, a phenomenon referred to as kinetic resistance (16). Because a far greater percentage of cancer cells than normal cells are in the growth cycle (as opposed to G0), drug treatment should favor the destruction of malignant cells over normal cells.
Growth Kinetics
According to the gompertzian model of tumor growth, only a fraction of tumor cells in a mass are actively proliferating.
The smaller the tumor, the greater is the proportion of proliferating cells. Chemotherapeutic agents kill proliferating cells and thus are most effective in eradicating smaller tumors. This line of reasoning has been successfully applied to the treatment of micrometastases, metastases that are present at diagnosis but are too small to be seen by standard imaging modalities. Local control of tumors is therefore sufficient therapy for fewer than 20% of patients with tumors, including Ewing’s sarcoma, rhabdomyosarcoma, osteosarcoma, and non-Hodgkin’s lymphoma. Addition of effective chemotherapy to local control has reduced the incidence of subsequent metastases and accounts for the marked increased cure rates that began in the 1970s (17).
The smaller the tumor, the greater is the proportion of proliferating cells. Chemotherapeutic agents kill proliferating cells and thus are most effective in eradicating smaller tumors. This line of reasoning has been successfully applied to the treatment of micrometastases, metastases that are present at diagnosis but are too small to be seen by standard imaging modalities. Local control of tumors is therefore sufficient therapy for fewer than 20% of patients with tumors, including Ewing’s sarcoma, rhabdomyosarcoma, osteosarcoma, and non-Hodgkin’s lymphoma. Addition of effective chemotherapy to local control has reduced the incidence of subsequent metastases and accounts for the marked increased cure rates that began in the 1970s (17).
The rationale for prolonged intermittent treatment with chemotherapy has its roots in the concept of kinetic resistance. Cells not cycling at the time of treatment are in G0 and therefore are likely to be resistant to the effects of growth cycle-specific chemotherapy. These cells can lay dormant for weeks or even months before reentering the cell cycle. Intermittent pulses of chemotherapy delivered over months increase the likelihood of catching these dormant cells as they reenter the growth cycle and are more susceptible to chemotherapy.
Combination Chemotherapy
Since the mid-1970s, oncologists have moved from simple, single-agent regimens of chemotherapy to more complex and intensive programs. The new regimens have evolved through clinical trials based not only on the biologic principles described earlier, but also on experience. Drug resistance can be attributable to many factors, such as the presence of a preexisting resistant clone or the development of a secondary mutation that confers drug resistance. The general strategies currently employed include combination chemotherapy, dose intensity, rescue after dose-intensive therapy, and potentiators of chemotherapy.
The use of multiple drugs for the treatment of pediatric malignancies is now standard. These drugs can be administered concomitantly, as in the four- to six-drug regimens employed for remission induction for ALL. They can also be administered sequentially, as in the alternating courses of doxorubicin/vincristine/cyclophosphamide and VP-16/ifosfamide for Ewing’s sarcoma. This approach has its origins in the Goldie-Coldman hypothesis. According to this hypothesis, proliferating cancer cells are at continued risk of undergoing mutations, and it is possible that some chance mutation will cause the cells to become resistant to certain types of chemotherapy. With a mutation rate of approximately 1 per 105 cells, it is statistically improbable that a tumor will develop resistance to more than one agent. Numerous clinical trials have substantiated the Goldie-Coldman hypothesis. The Dana Farber Cancer Institute protocol for childhood ALL illustrates the dramatic improvements in cure rates with combination chemotherapy (18). In the 1970s, standard induction treatment for ALL was vincristine and prednisone. Remission was easily achieved, but many relapsed. The addition of doxorubicin increased the remission rate that was already 90% slightly, but had a significant impact on the event-free survival rate, increasing it from 38% to 69%. When L-asparaginase was added to the three drugs, event-free survival climbed even higher to 79%. Now, with a six-drug induction therapy, remission rates for ALL are 99%, and relapse-free survival rates are as high as 89%.
The following principles guide the choices of agents for these combination regimens: (1) the drugs should have documented activity against a given tumor, (2) the drugs should have differing mechanisms of action, (3) there should be as little toxicity overlap as possible, and (4) the drugs should have different patterns of resistance.
Dose Intensity
Many antineoplastic drugs have been shown to have steep dose–response curves—that is, the higher the dose, the greater the tumor cell kill. Clinical studies have demonstrated that increased dose intensity is associated with increased survival (17,19,20). Dose intensity can take the form of higher doses of a drug or more frequent delivery of standard doses of the drug. The importance of the latter is illustrated in a review of children who received varying doses of chemotherapy for ALL and osteosarcoma. Children who received less than 75% of the intended doses of chemotherapy did not fare as well as those who received greater than 75% (19,20).
One method of safely increasing dose intensity is to provide a “rescue” from the toxicity of high-dose chemotherapy after allowing the high doses of the drug to render their antitumor effect. Perhaps the most dramatic example of this is autologous or allogeneic bone marrow transplantation. Such an approach is generally reserved for the patient whose malignancy is not treatable by standard chemotherapy. In this instance, the patient’s (or a suitable donor’s) bone marrow is harvested and saved. The patient then receives massive doses of chemotherapy (sometimes with total-body irradiation), which ablates the remaining bone marrow. The patient is then rescued from bone marrow aplasia by reinfusion of the patient’s own (or the donor’s) bone marrow.
High-dose local therapy also increases dose intensity. Examples include intrathecal chemotherapy for cerebrospinal fluid tumors, intraperitoneal therapy for ovarian cancer, and intraarterial administration of chemotherapy in the region of the tumor. Another means of rescue is the use of leucovorin after administration of high doses of methotrexate. Methotrexate inhibits the enzyme dihydrofolate reductase that plays a critical role in the synthesis of DNA bases. Leucovorin (tetrahydrofolate) is the product of the enzyme; thus, once leucovorin has been
given, the enzyme is bypassed, DNA bases are again synthesized, and the severe myelosuppression and mucositis caused by methotrexate are avoided. Finally, safe administration of ifosfamide and high doses of cyclophosphamide were made possible by the concomitant administration of mesna, a compound that prevents cyclophosphamide and ifosfamide-induced hemorrhagic cystitis.
given, the enzyme is bypassed, DNA bases are again synthesized, and the severe myelosuppression and mucositis caused by methotrexate are avoided. Finally, safe administration of ifosfamide and high doses of cyclophosphamide were made possible by the concomitant administration of mesna, a compound that prevents cyclophosphamide and ifosfamide-induced hemorrhagic cystitis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


