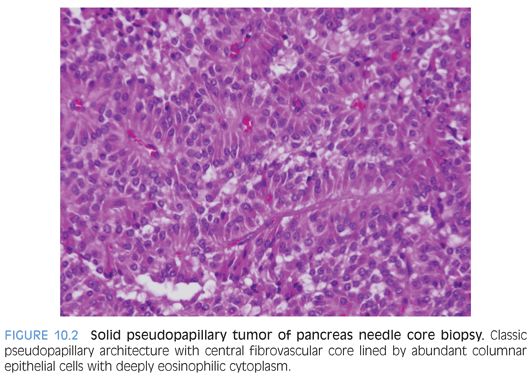
Solid pseudopapillary tumor of pancreas (SPN) is a low-grade malignancy with epithelial differentiation, most common in adolescents and young women.4–6 The histopathologic features vary from solid to pseudopapillary to cystic areas (Fig. 10.2 and eFig. 10.2). The tumor cells possess eosinophilic cytoplasm with grooved to indented to reniform nuclei with small to inconspicuous nucleoli. Deeply eosinophilic hyaline globules are seen both in the cytoplasm as well as extracellularly. Cholesterol clefts, foamy macrophages, calcifications, infarction, hemorrhage, and hemosiderin deposition may be present. IHC profile includes nuclear β-catenin, CD99 Golgi dot-like pattern, progesterone receptors, CD10, cyclin D1, α1-antitrypsin, FLI1, and CD117, with variable reactivity with synaptophysin, CD56, and NSE.6 Claudins 5 and 7 distinguish solid pseudopapillary tumors from PCB, acinar clear cell carcinoma, and endocrine tumors of the pancreas.6 These tumors are also distinct from ductal adenocarcinoma.
Serous cystadenoma (SC) in children is extremely rare, most likely derived from centroacinar cell origin.7 The cysts are lined by a single layer of low columnar to cuboidal cells to flattened epithelial cells with clear to pale cytoplasm lacking mucinogenic features and small round to ovoid bland nuclei (eFig. 10.3). The differential diagnosis includes pseudocyst, congenital cyst, acquired cyst, and cystadenoma, either serous or mucinous.
Congenital cysts of pancreas are rare, and may be diagnosed in utero or perinatally. It appears that the sequestered segments of a primitive secretory duct system gives rise to multiple microscopic and macroscopic cysts lined by cuboidal to low columnar epithelial cells (Fig. 10.3 and eFig. 10.4). The identification of congenital multicystic or polycystic pancreatic lesions warrants evaluation for other anomalies and possible associated syndromes. Unilocular congenital (dysgenetic) pancreatic cysts may occur and are thought to be derived from anomalous development of the pancreatic duct system. The cyst wall is lined by cuboidal to low columnar epithelium. Fibrous and inflammation may be present with loss of the epithelial lining.

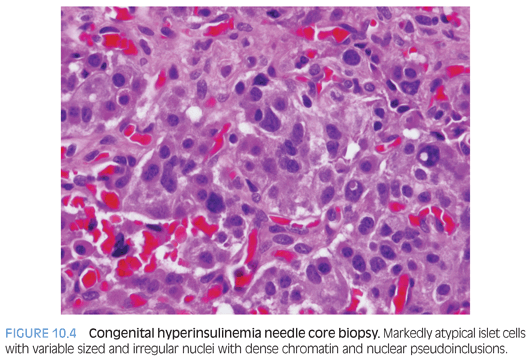
Congenital hyperinsulinism (HI) is characterized by profound hypoglycemia due to unregulated insulin secretion.8,9 Diffuse form of HI has confluent or partially confluent clusters of islet cells in the pancreas, implying that all beta cells throughout the entire pancreas are affected. The beta cells have abundant cytoplasm with abnormal nuclei that are three to four times the size of acinar nuclei (sometimes termed giant islet cells). The focal form is usually restricted to a small area within the pancreas (2.5 to 7.5 mm diameter). The beta cells also have large amounts of cytoplasm, irregular angulated large nuclei (three to five times the size of acinar nuclei). The focal lesion tends to be multilobular and may have satellite lesions in proximity to the more defined lesional area.
A preoperative biopsy alone (Fig. 10.4 and eFig. 10.5) will not provide a definitive answer to whether the diffuse or focal form of HI exists, although it may provide assistance in localization of a focal lesion and provide an initial “starting point” for surgical excision. Prior to surgery, it is important that all modulating medications be discontinued (5 days for diazoxide, 2 days for octreotide) because of interference with histopathologic interpretation.8,9 Biopsies of the tail, body, and head are performed to determine if HI is of the focal or diffuse form. Surgical excision of the focal form of HI with frozen section margins to assess for complete excision is necessary to affect a “cure.”
ADRENAL GLAND BIOPSIES IN PEDIATRICS
Most adrenal glands examined in surgical pathology are resection specimens with biopsies being exceedingly rare. The more common scenario is a biopsy of a metastatic tumor from the adrenal gland, typically a neuroblastoma. The sites of metastases include skin, liver, bone marrow, and lymph nodes. Diagnostic workup is similar to that performed for a primary resection because the biology and morphology tends not to change in the metastases. Other rare tumors occurring in children include paraganglioma and adrenal cortical adenoma and carcinoma (eFigs. 10.6 and 10.7), which are not discussed here.
Neuroblastoma Family of Tumors
Neuroblastoma is a family of tumors (Figs. 10.5 to 10.7 and eFigs. 10.8 to 10.13) that spans a spectrum from a mature benign form (ganglioneuroma) to a partial mature ganglionic and Schwannian stroma–rich tumor with residual neuroblasts (ganglioneuroblastoma) to neuroblast-rich and Schwannian stroma–poor to Schwannian stroma–absent tumors (neuroblastoma).10–13 Tumors arise in the location of the sympathetic chain, most commonly from the adrenal medulla, sympathetic paravertebral ganglia, and sympathetic paraganglia, such as the organ of Zuckerkandl. In congenital forms, hydrops fetalis and blue-red cutaneous lesions (blueberry muffin baby) may be present. Although a palpable mass is present in two-thirds of children, clinical presentation varies from an asymptomatic mass to locally invasive disease to widespread metastatic disease. Some tumors mature spontaneously into ganglion cells with Schwannian stroma (ganglioneuroma).
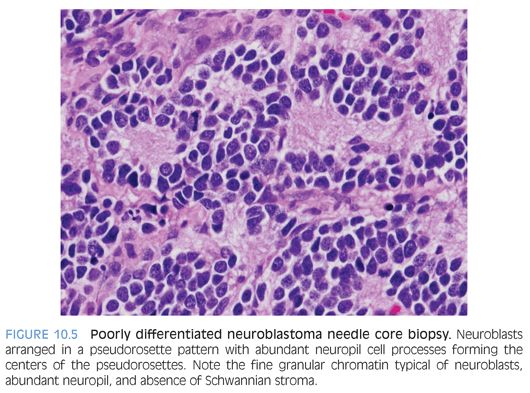
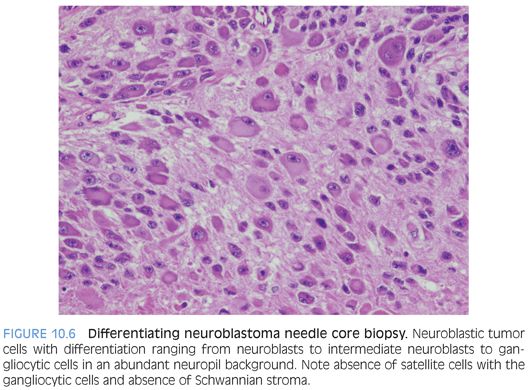
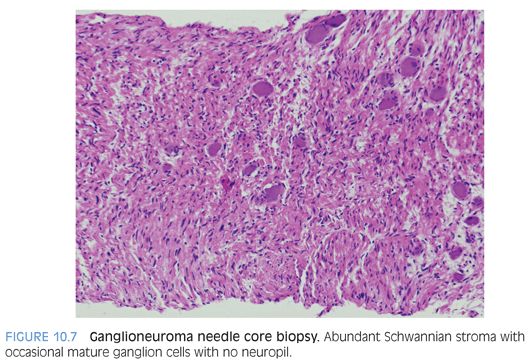
Neuroblastic tumors are composed of three classes, based on differentiation of the neuroblastic cells and proportion of Schwannian stroma (eTable 10.1).10–13 The Schwannian stroma–poor neuroblastoma class is further subclassified as undifferentiated, poorly differentiated, and differentiating neuroblastoma. Undifferentiated neuroblastoma (Fig. 10.8) is a small round cell tumor without evidence of typical neuroblastic differentiation. The tumor cells have a high nuclear to cytoplasm ratio and lack neuropil cell processes. There is no Schwannian differentiation. The nuclear morphology may have fine granular chromatin and distinct nucleoli. The differential diagnosis includes all embryonal small round cell malignancies of childhood. Tumor location and elevated urinary catecholamine metabolites (vanillylmandelic acid, homovanillic acid) may provide evidence for a neuroblastic tumor.




IHC, electron microscopy, and cytogenetics are necessary to establish a diagnosis in undifferentiated neuroblastomas. Neuroblasts immunoreact with antibodies to NB84 (Fig. 10.8), PGP9.5, NSE, chromogranin A, synaptophysin, tyrosine hydroxylase, CD56, and GD2. Neuroblasts are negative for vimentin, desmin, myogenin, low-molecular-weight cytokeratins, Wilms tumor 1 (WT1), epithelial membrane antigen (EMA), CD45, and CD99. Electron microscopy of an undifferentiated neuroblastoma demonstrates neuroblasts with fine neurite cytoplasmic processes and dense core neurosecretory granules.
Neuroblastic tumors are assigned a favorable or unfavorable histology (eTable 10.1). This is based on appropriate classification and subclassification of the tumor, determining the mitotic–karyorrhexis index (MKI) for the tumor, and the patient’s age. Neuroblastomas are classified as low, intermediate, or high risk based on certain biologic and clinical factors (eTable 10.1). Histopathologic features have an impact on 5-year overall survival, which is also significantly affected by MKI, stage of disease, favorable verus unfavorable histology, MYCN amplification, and patient age.10–13
Genetic mutations associated with sporadic and familial inherited neuroblastomas have been identified.14 Activating anaplastic lymphoma kinase (ALK) (2p23-24) gene mutations account for most of the hereditary neuroblastomas. Somatic mutations in ALK have been reported in 5% to 15% of sporadic neuroblastomas. ALK-inhibitor medications are currently available for targeted therapy. The PHOX2B (4p12) gene (autonomic nervous system development regulator) plays a major role in tumorigenesis in both hereditary and sporadic neuroblastomas. Known genetic abnormalities with neuroblastoma include MYCN amplification, loss of tumor suppressor genes at 1p and 3p, and deletion of the cell cycle regulator cyclin-dependent kinase inhibitor 2A (CDKN2A) on chromosome 9. Methylation silencing of gene function is a common mechanism in neuroblastoma development. Gene methylation in apoptosis (caspase 8, TMS1) and cell cycle (CDKN2A, CCND2, SFN) regulation participate in neuroblastoma development. Dysregulation of retinoic acid receptor genes and cell adhesion molecules (cadherin 1) participate in the neoplastic process. Small nonencoding RNAs (microRNA [miRNA]) act as negative regulators, affecting MYCN amplification in both sporadic and familial neuroblastomas.
Certain chromosomal aberrations (translocations, amplifications, deletions, regional chromosomal losses and gains) are associated with aggressive neuroblastomas. In contrast, whole chromosomal gains (hyperdiploidy, chromosomal duplications) are associated with “benign” or less aggressive neuroblastomas. Based on genome-wide association studies, it has been possible to stratify neuroblastomas and relate these to established low-, intermediate-, and high-risk categories and the very favorable stage 4S category. Genome-wide and whole-exome association studies may determine treatment, prognosis, event-free survival, and overall survival of individual patients in the near future.
THYROID BIOPSIES IN PEDIATRICS
The prevalence of palpable thyroid nodules in children is approximately 1.5% in comparison to 4% to 8% in adults.15,16 These are particularly concerning in children because malignancy is identified in up to 50% (only 5% to 10% in adults) and they also have an increased prevalence of lymph node and distant site involvement. Over the past decade, evaluation of thyroid nodules in children has progressed from clinical and radiologic evaluation only prior to surgical intervention to FNA and fine needle core biopsy (FNB) of lesions.16 FNAs are unsatisfactory in 9%, benign in 72%, suspicious for malignancy in 12%, and malignant in 6%. With those children undergoing nodule resection, lesions with a benign FNA diagnosis proved in 98% of cases to be multinodular goiters or follicular adenomas. Nodules considered suspicious on FNA proved to be malignant in 20%; atypical adenomas with papillary features in 16%; and multinodular goiter, follicular adenoma, and autoimmune thyroiditis in 64%. Of those nodules considered malignant on FNA, the majority were papillary thyroid carcinoma (87%), follicular carcinoma (6%), and medullary carcinoma (6%) when resected. In a meta-analysis of pediatric thyroid FNA studies, FNA sensitivity of 94% and specificity of 81% were found. The accuracy, positive predictive value, and negative predictive value were 84%, 55%, and 98%, respectively.
Similar findings have been found with children undergoing FNB at a large pediatric hospital.16 FNBs were found to be nondiagnostic in 13%, benign in 24%, atypical in 37%, and malignant in 26%. Sensitivity and specificity for diagnosis of follicular neoplasms were 0.87 and 0.75, respectively. Sensitivity and specificity for diagnosis of papillary thyroid carcinoma and follicular neoplasm were 0.85 and 0.63, respectively. The advantage to performing an FNB is the ability to have tissue cores available for several purposes. Tissue can be submitted for cytogenetics and molecular genetic studies, including gene rearrangement (translocation detection by reverse transcriptase–polymerase chain reaction [RT-PCR]), cytologic (touch) imprints may be made from the cores for FISH and CISH, tissue may be cryopreserved for possible whole-genome assay and whole-exome studies, and tissue cores may be routinely processed allowing for routine IHC staining for diagnostic and prognostic markers.
At pediatric hospitals, where children have been referred for partial or complete thyroidectomy, there appears to be an overrepresentation of malignant diagnoses.15,16 Malignancy was reported to represent 36% of cases, with most being papillary thyroid carcinoma followed by follicular carcinoma, medullary carcinoma, and a single case of primary Ewing sarcoma. The most common benign diagnosis was follicular adenoma.
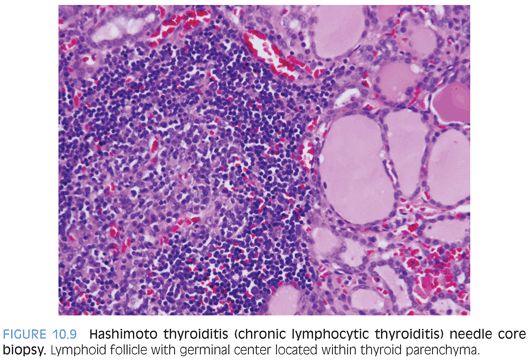
Hashimoto thyroiditis (chronic lymphocytic thyroiditis) is an autoimmune disease that is characterized by circulating antithyroid peroxidase, antithyroid microsomal, and antithyroglobulin antibodies, with activation of CD4 T lymphocytes directed toward follicular cell antigens and recruitment of cytotoxic CD8 T lymphocytes. This inflammatory process leads to diffuse infiltration of the thyroid with eventual loss of parenchymal cells and replacement with fibrous tissue, resulting in hypothyroidism. The thyroid gland is tender on palpation and has a granular to pebbly texture. This condition is typically rare in children. About one-third of children have spontaneous resolution with return to normal thyroid function and loss of antibodies. In adolescents, Hashimoto thyroiditis may represent 40% of thyroid goiters. This disease may be associated with other autoimmune diseases (systemic lupus erythematosus, Sjögren syndrome, immunoglobulin G [IgG]4–type diseases) and other diseases (diabetes, Addison disease, myasthenia gravis, pernicious anemia, thrombocytopenia purpura, hepatitis). The affected individuals may complain of neck tightness but rarely of discomfort. Females are more commonly affected. The granular to pebbly thyroid gland may be tender on palpation. Macroscopically, the thyroid is diffusely markedly enlarged (two to three times increase, weight up to 200 g) with possible adherence to extrathyroidal soft tissue and muscle secondary to the chronic inflammatory process. FNB shows a diffuse lymphoplasmacytic inflammatory infiltrate with readily identified lymphoid follicles with germinal centers (Fig. 10.9 and eFig. 10.14). The inflammatory infiltrate is composed of B and T lymphocytes and plasma cells with no clonality on kappa and light chain immunostaining. There are no BRAF mutations. In rare cases, B-cell lymphoma may develop in the background of Hashimoto thyroiditis.