Normal and Abnormal Sexual Development
 |
Abnormalities of sexual differentiation are seen infrequently in an individual clinician’s practice. However, few physicians have not been challenged at least once by a newborn with ambiguous genitalia or by a young woman with primary amenorrhea. Traditional classifications for disorders of sexual differentiation have been confusing, but advances in reproductive science have helped to define their causes and to provide the foundation for a logical and efficient approach to diagnosis.
This chapter first considers the processes involved in normal sexual differentiation, to provide a basis for understanding the various types and causes of abnormal development. Some subjects are discussed in other chapters, but also are included here, for clarity and completeness. The fundamental theme is that disorders of sexual development result primarily from abnormalities in the amount or action of androgens—from excess androgen in females and from too little androgen in males.
Normal Sexual Differentiation
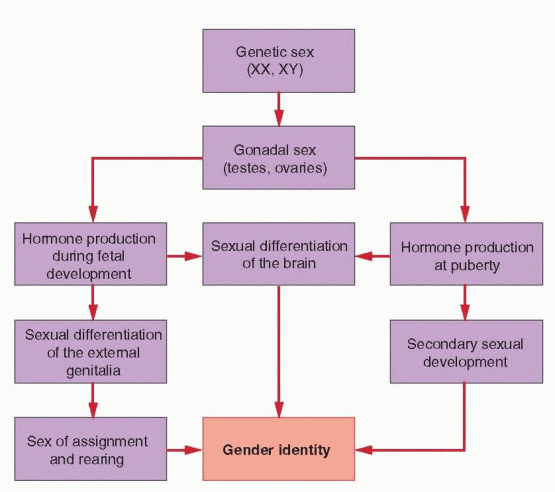
The gender identity of a person (whether an individual identifies as a male or a female) is determined by their genetic, gonadal, and phenotypic sex and also is influenced by their
environment. Genetic or chromosomal sex is defined by the sex chromosomes, typically XX or XY. Gonadal sex is defined by the direction of gonadal differentiation, into ovaries or testes. Phenotypic sex is defined primarily by the appearance of the external genitalia and the secondary sexual characteristics that develop at puberty. Gender identity includes all behavior having any sexual connotation, such as body gestures and mannerisms, habits of speech, recreational preferences, and content of dreams. Sexual expression, both homosexual and heterosexual, reflects the sum of all sexual influences on the individual, both prenatal and postnatal, the latter referring to the role assigned by society in accordance with the individual’s phenotype and behavior.
environment. Genetic or chromosomal sex is defined by the sex chromosomes, typically XX or XY. Gonadal sex is defined by the direction of gonadal differentiation, into ovaries or testes. Phenotypic sex is defined primarily by the appearance of the external genitalia and the secondary sexual characteristics that develop at puberty. Gender identity includes all behavior having any sexual connotation, such as body gestures and mannerisms, habits of speech, recreational preferences, and content of dreams. Sexual expression, both homosexual and heterosexual, reflects the sum of all sexual influences on the individual, both prenatal and postnatal, the latter referring to the role assigned by society in accordance with the individual’s phenotype and behavior.
Normal sexual differentiation involves a sequence of related processes that begins with genetic or chromosomal sex, as established at the time of fertilization.1 Gonadal sex is determined next; directed by the genetic sex, the indifferent gonads differentiate into ovaries or testes. In turn, gonadal sex controls the hormonal environment of the embryo, which directs the development of the internal and external genitalia. The processes involved in sexual differentiation of the embryonic brain are less clear, but may involve mechanisms similar to those controlling differentiation of the external genitalia. The inductive influences of hormones on the developing central nervous system (CNS) ultimately may determine the patterns of hormone secretion and sexual behavior in the adult.2,3,4,5,6 and 7
Although the mechanisms that govern sex differentiation are not yet entirely clear, our understanding of the molecular processes involved has advanced significantly in recent years. Current concepts are summarized here, beginning with the genetics of sex determination, followed by germ cell sex differentiation, gonadal differentiation, and development of the internal and external genitalia.
 |
Genetics of Sex Determination
Both the X and the Y chromosomes appear to have evolved from autosomal ancestors over a period of 300 million years.8 Most of the ancestral genes on the Y chromosome have been lost in the process, leaving only a limited number of currently active genes. A great many
genes are involved in translating the sex chromosome composition of the embryo and in directing the differentiation of the gonadal somatic cells,9,10 and 11 but sex determination depends primarily on the presence or absence of a Y chromosome.
genes are involved in translating the sex chromosome composition of the embryo and in directing the differentiation of the gonadal somatic cells,9,10 and 11 but sex determination depends primarily on the presence or absence of a Y chromosome.
In females, the identical pair of X chromosomes aligns and recombines along its entire length during meiosis, like the autosomes. In males, homology between the X and Y chromosomes is limited to two small regions located at the very distal ends of the short and long arms of the Y. The “pseudoautosomal” region comprises only approximately 5% of the entire Y chromosome and is the only region that normally pairs and recombines during meiosis.10, 12 Most of the remaining 95% of the Y chromosome is unique to the male, containing multiple copies of genes expressed specifically in the testis and encoding proteins with specialized functions.8 A single copy of the one gene most critical to testis differentiation, SRY (Sex-determining Region on Y), is located on the distal short arm of the Y (Yp11.3), immediately adjacent to the pseudoautosomal region.13
Most of what is known about the genetic basis for sexual differentiation derives from studies of mutations in the mouse and human associated with varying degrees of “sex reversal,” conditions in which the chromosomal sex does not correlate with the gonadal or phenotypic sex. In humans, 46,XX male sex reversal occurs when pairing between the X and Y chromosomes during male meiosis extends abnormally into adjacent non-homologous regions, allowing inappropriate recombination and transfer of Y-specific DNA onto the X chromosome. Careful analysis of four XX males having a very small piece of translocated Y DNA (60 kb)14 prompted a search for highly conserved sequences within that region, which led to discovery of the SRY gene.13 The identification of SRY mutations in three XY females supported the hypothesis that SRY was the critical and long sought “testis determining factor,”15, 16 but proof derived ultimately from studies in the mouse. First, a deletion in Sry (by convention, mouse genes are designated by small case letters) was identified in a line of XY female mice.17 Second, Sry gene expression in the genital ridge was observed just at the time of testis differentiation.18 Third, transgenic XX mice carrying Sry develop as males.19 SRY now is generally established as the primary genetic signal determining the direction of gonadal differentiation in mammals.10,20 However, XX hermaphrodites having ovotestes but not SRY have been described and only a small proportion of phenotypic females with XY gonadal dysgenesis (Swyer syndrome) harbor SRY mutations. These observations indicate clearly that sex determination and sex reversal involve genes other than SRY.21
Although the mechanisms that regulate SRY expression are still unclear, the nuclear receptor SF1 (Steroidogenic Factor 1) has emerged as a likely and important activator. In the mouse, Sf1 binds to and activates the Sry promoter,22 and heterozygous mutations in the Sf1 gene (resulting in haploinsufficiency) produce XY female sex reversal.23,24 and 25 In humans, SF1 haploinsufficiency is a known cause of XY female sex reversal,26 and an SF1 polymorphism that reduces transactivation function by approximately 20% is recognized as a susceptibility factor for the development of micropenis and cryptorchidism.27, 28 Evidence indicates that splice variants of Wt1 (Wilms tumor 1) and GATA4 (GATA binding protein 4) also may be involved in the regulation of Sry expression; both are transcription factors containing zincfinger motifs that can interact and synergistically activate the promoter of human SRY.29 WT1 mutations are associated with gonadal dysgenesis and ambiguous genitalia in males.30
The sequence of molecular events involved in testis differentiation is not completely understood, but SRY appears to activate a number of other genes that promote testis development.31 The 204 amino acid protein product of SRY (SRY) contains a 79 amino acid domain very similar to that in a recognized family of transcription factors known as the high mobility group (HMG), which bind to DNA and regulate gene transcription. Members of the related SRY HMG box (SOX) protein family of transcription factors play a crucial role in the cascade of events that drives testis differentiation, and most of the SRY point mutations identified in sex-reversed patients translate to abnormalities in the amino acid sequence of SOX proteins.32
Substantial evidence now indicates that SOX9 is the most likely SRY target gene. In mice, Sox9 expression is dramatically up-regulated soon after Sry expression begins in XY gonads but down-regulated in XX gonads,33 and cell-fate mapping experiments have found that Sox9-positive Sertoli cells derive exclusively from Sry-positive gonadal somatic cells.34 XY mouse embryos having a targeted deletion of Sox9 develop ovaries,35, 36 and transgenic activation of Sox9 expression induces male development in XX embryos.10 In humans, heterozygous mutations in SOX9 (resulting in haploinsufficiency) cause a skeletal malformation syndrome (campomelic dysplasia) in which most affected XY patients exhibit female sex reversal, and SOX9 duplication (resulting in overexpression) is the only known autosomal cause of XX male sex reversal.32
The developmental consequences of activating and inactivating mutations in Sox9 resemble those of similar mutations in Sry, implying not only that Sox9 is required for testis differentiation, but also that Sry activation of Sox9 may be all that is necessary to activate other genes important to testis development, such as Fgf9 (fibroblast growth factor 9), and to repress genes that induce ovary development, such as Wnt4 (a member of the wingless family of genes), Rspo1 (R–spondin 1), Dax1 (dosage-sensitive sex reversal, adrenal hypoplasia critical region, on chromosome X, gene 1), and Foxl2 (forkhead box L2).32 DAX1 is a nuclear transcription factor normally upregulated in the ovary and repressed by SOX9, but DAX1 duplication (resulting in overexpression) can repress SRY (directly, or indirectly by inhibiting SF1) and cause XY female sex reversal.37, 38 SOX9 probably is the one most important factor regulating the activity of genes involved in Sertoli cell differentiation, and evidence suggests that SOX9 drives the process via feed forward loops that up-regulate its own expression. Sox9 stimulates Sf1 expression, binds to the same enhancer as Sry (after Sry expression has ended), and also stimulates Fgf9 expression in nascent Sertoli cells, all of which up-regulate Sox9 expression and combine to maintain high levels of Sox9 activity.10, 31, 32 Although a great many genes are involved in testis differentiation, virtually all male-to-female sex reversal in mice and in humans can be explained ultimately, directly or indirectly, by the failure to generate sufficient levels of SOX9 to promote the positive-feedback loops that maintain its expression.
Fgf9 appears particularly critical for maintaining the levels of Sox9 expression required to induce testis differentiation. Both Fgf9 and Sox9 are expressed at low levels in bipotential
XX and XY gonads, but Fgf9 expression is lost in XX and amplified in XY gonads soon after Sry is expressed.39 Deletion of Fgf9 does not prevent initial expression of Sry or Sox9 in Sertoli cell precursors, but Sox9 expression is a prerequisite for Fgf9 expression, and without it, Sox9 expression cannot be sustained.40 Fgf9 also appears to actively repress genes that promote ovary differentiation, such as Wnt4.39
XX and XY gonads, but Fgf9 expression is lost in XX and amplified in XY gonads soon after Sry is expressed.39 Deletion of Fgf9 does not prevent initial expression of Sry or Sox9 in Sertoli cell precursors, but Sox9 expression is a prerequisite for Fgf9 expression, and without it, Sox9 expression cannot be sustained.40 Fgf9 also appears to actively repress genes that promote ovary differentiation, such as Wnt4.39
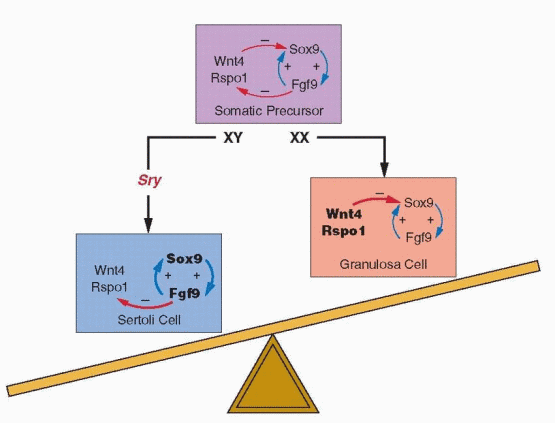 |
Whereas ovarian differentiation has long been considered the “default” pathway of sex determination—the automatic result in the absence of a testis determining factor—recent evidence challenges that traditional concept. In mice, inactivating mutations in genes such as Wnt4,39, 41 Rspo1,42,43 and 44 and Foxl245,46 and 47 result in partial or complete XX male sex reversal, and activating mutations in β-catenin or Dax1 result in XY female sex reversal.32, 48, 49 Rspo1 is required for Wnt4 expression and activates β-catenin, which, like Foxl2, downregulates Sox9 expression.21 Dax1 acts as a dominant-negative regulator of transcription of other nuclear receptors, including SF1, and thus may repress Sry expression.32 Taken together, these observations suggest strongly that ovarian development results from the active repression of one or more genes in the testis pathway, rather than from a developmental default mechanism.
It now appears that both testis and ovary differentiation require dominantly acting genes, with SRY inducing testis development via up-regulation of SOX9, and with other genes, primarily WNT4 and RSPO1, teaming to promote ovary development via repression of SOX9. The new concept views the fate of the bipotential gonad as balanced between opposing forces and SRY as the key factor. In XY gonads, SRY induces SOX9 and tips differentiation toward testis development, and in XX gonads lacking SRY, other genes combine to repress SOX9 and promote ovary development.21, 50
Germ Cell Sex Differentiation
In human embryos, gonadal development begins during the fifth week of gestation as a protuberance overlying the mesonephric ducts, known as the genital or gonadal ridge. The primordial germ cells do not arise within but migrate into the developing gonads between 4 and 6 weeks gestation, proliferating as they go. At least in the mouse, their survival during migration appears to depend on an interaction between the cell surface tyrosine kinase receptor, c-KIT, and a ligand produced by surrounding tissues, called stem cell factor.51 At this stage of development, the gonads are identical in males and females, indifferent and bipotential, capable of differentiating into either testes or ovaries in response to inductive signals. Although germ cells do not induce gonadal development, they play a more active role in females than in males. In the genetic or pharmacologically induced absence of germ cells, testis cords (the embryonic precursor to seminiferous tubules in the adult testis) can develop, but in females, ovary differentiation fails altogether;52, 53 somatic cells aggregate but deteriorate, leaving only stromal tissue, and ultimately, a fibrous streak. After arrival in the nascent gonads, germ cell differentiation into male (prospermatogonia) or female (oogonia) depends on the sex of the gonadal somatic cells and on signals in the surrounding environment rather than on the chromosomal sex of the germ cells themselves. In XY/XX mouse chimeras, XY primordial germ cells can develop as oogonia in female embryos, and XX germ cells as prospermatogonia in male embryos.54
It is not yet clear whether the signaling molecules that mediate germ cell sex determination act in the developing testis to inhibit meiosis or in the developing ovary to induce meiosis, what those signaling molecules may be, and whether they act directly on the germ cells themselves, or indirectly via actions on gonadal somatic cells.31 Recent studies in mice aimed at identifying molecular candidates for the putative meiosis inducing or inhibiting factors have focused attention on retinoic acid, which is produced in the mesonephros.
Whereas retinoic acid treatment induces primordial germ cells in male gonadal explant cultures to express Stra8, Scp3, and Dmc1 (meiosis marker genes), germ cells in female gonadal explants treated with a retinoic acid inhibitor continue to express Oct4 (a marker for pluripotent cells).55 Moreover, Sertoli cells, which surround the germ cells in the developing testis cords, express Cyp26B1, a gene encoding an enzyme (CYP26B1) that metabolizes retinoic acid.56 Taken together, these observations suggest that local levels of retinoic acid may regulate germ cell differentiation in the developing gonad, with retinoic acid diffusing from the adjacent mesonephros acting as the functional meiosis inducing factor in female germ cells, and with CYP26B1 produced by Sertoli cells in the developing testis cords acting as the functional meiosis inhibiting factor in male germ cells.10 Alternatively, or in addition, Sertoli cells may secrete a specific meiosis inhibiting factor, with one likely downstream target being Nanos2, a gene expressed exclusively in male germ cells.31, 57
Whereas retinoic acid treatment induces primordial germ cells in male gonadal explant cultures to express Stra8, Scp3, and Dmc1 (meiosis marker genes), germ cells in female gonadal explants treated with a retinoic acid inhibitor continue to express Oct4 (a marker for pluripotent cells).55 Moreover, Sertoli cells, which surround the germ cells in the developing testis cords, express Cyp26B1, a gene encoding an enzyme (CYP26B1) that metabolizes retinoic acid.56 Taken together, these observations suggest that local levels of retinoic acid may regulate germ cell differentiation in the developing gonad, with retinoic acid diffusing from the adjacent mesonephros acting as the functional meiosis inducing factor in female germ cells, and with CYP26B1 produced by Sertoli cells in the developing testis cords acting as the functional meiosis inhibiting factor in male germ cells.10 Alternatively, or in addition, Sertoli cells may secrete a specific meiosis inhibiting factor, with one likely downstream target being Nanos2, a gene expressed exclusively in male germ cells.31, 57
In the male, the primordial germ cells become incorporated into the developing testis cords and enter mitotic arrest as prospermatogonia, resuming proliferation soon after birth. In the female, the primordial germ cells (oogonia) continue to proliferate by mitosis somewhat longer, reaching a peak of 5-7 million by 20 weeks of gestation. However, only some enter meiosis and become primary oocytes, arresting in diplotene of the first meiotic prophase, and become surrounded by a single layer of flattened pregranulosa cells, forming primordial follicles. Those that are not incorporated into primordial follicles degenerate via apoptosis and, by birth, only approximately 1-2 million germ cells remain. The signals for programmed cell death are unknown but seem likely to involve some form of intercellular communication between the primary oocyte and surrounding pregranulosa cells.
Whereas male germ cells proliferate continuously, the traditional dogma has held that female germ cells proliferate only during embryogenesis and, therefore, that females are born with a finite number of primordial follicles that are steadily depleted and cannot be replenished. However, that dogma has been challenged by studies suggesting that germ line stem cells reside within the bone marrow and may replenish the ovary with new oocytes,58, 59 stimulating a vigorous scientific debate,60,61,62,63,64,65 and 66 which continues. Whether or not it occurs normally, the demonstration that mice sterilized by chemotherapy can produce offspring derived from intra-ovarian transplants of germ line stem cells isolated from neonatal or adult ovaries argues that germ line stem cells reside in the ovary and that postnatal oogenesis is possible.67
Testis Differentiation and Development
The current model for testis differentiation and development, based primarily on studies in mice, envisions a sequence of events that begins with the formation of the genital ridge, first recognized as a thickening underlying the coelomic epithelium adjacent to the mesonephros. Primordial germ cells migrate into the genital ridge, along with proliferating coelomic epithelial cells, which express Sf1. A portion of the epithelial daughter cells expresses Sry to become Sertoli cell precursors, the first cell type to differentiate and the only cell type in the developing testis that expresses Sry. The subset of somatic cells expressing Sry immediately also begins to express Sox9, a reliable marker for developing Sertoli cells. In turn, Sox9-positive Sertoli cell precursors secrete other paracrine signaling molecules such as Fgf9 and prostaglandin D2 (PGD2), which also play important roles in testis differentiation. Ffg9 reinforces Sox9 expression and induces neighboring cells to proliferate, thereby increasing the generation of supporting cell precursors that are able to express Sry. PGD2 can induce even Sry-negative cells to express Sox9 and to differentiate into Sertoli cells.34 Together, Fgf9 and PGD2 help to maintain Sox9 levels and to ensure a
sufficient number of Sertoli cells to form a testis. Once the number of Sox9-positive cells reaches a critical threshold, Sox9 represses Sry expression.
sufficient number of Sertoli cells to form a testis. Once the number of Sox9-positive cells reaches a critical threshold, Sox9 represses Sry expression.
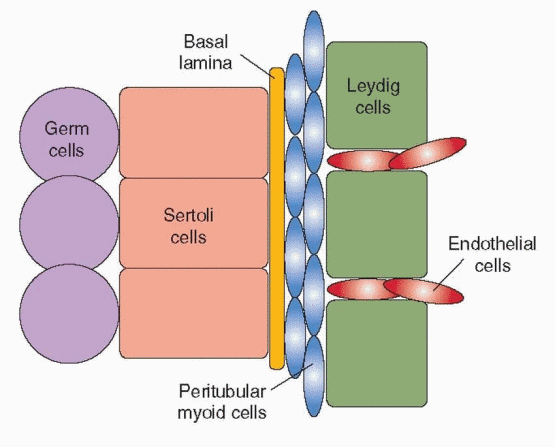
Under the control of Sry, Sertoli cells also secrete a factor that induces a migration of cells from the adjacent mesonephros. The developing testis enlarges rapidly with the influx of migrating cells, which differentiate into endothelial cells and Leydig cells upon their arrival in the developing gonad.10 Male-specific peritubular myoid cells appear to differentiate from cells already within the gonad, flattening and surrounding aggregates of Sertoli cells that organize in layers around clusters of primordial germ cells.50 The peritubular myoid cells thus help to form the testis cords, later serving to promote the movement of sperm through the seminiferous tubules in the adult testis. Together, the Sertoli cells and peritubular myoid cells induce the development of a basal lamina between them, separating the testis cords from the interstitial tissue. The steroidogenic Leydig cells differentiate within the interstitium, in close proximity to developing blood vessels that derive from endothelial cell precursors. Endothelial cell migration from the mesonephros is specific to the male and required for development of an arterial network that extends throughout the interstitium but not into the testis cords.50
 |
Ovary Differentiation and Development
In females lacking a Y chromosome and SRY, the bipotential gonad begins to differentiate into an ovary about 2 weeks later than testis development begins in the male. Normal ovarian differentiation requires the presence of germ cells; in their absence, the gonadal somatic cells fail to differentiate, indicating some form of communication between germ cells and somatic cells.53 Wnt4 and Rspo1 are two genes that play an important role in ovarian differentiation; XX mice with targeted deletions of either gene develop ovotestes containing sex cords and functional Leydig cells.43 Wnt4 expression is female-specific, suppresses the migration of mesonephric cells as occurs in the developing testis, and is dependent on Rspo1.41, 43 Rspo1 is specifically upregulated in XX somatic cells from the earliest stages of gonadal differentiation and encodes a secreted protein that, like Wnt4, activates the β-catenin signaling pathway in somatic cells, resulting in a loss of cell-cell adhesion between female germ cells, which is a prerequisite for their entry into meiosis.43 Consequently, directly or indirectly, Rspo1 regulates female germ cell and ovarian differentiation, by promoting events required for initiation of meiosis, inhibiting migration of mesonephric cells via Wnt4 expression, and by down-regulating Sox9, which drives testis
differentiation. Thus, whereas testis differentiation is directed by somatic cells, ovary differentiation requires communication between somatic cells and germ cells.68
differentiation. Thus, whereas testis differentiation is directed by somatic cells, ovary differentiation requires communication between somatic cells and germ cells.68
Gradually, the developing ovary becomes organized into an outer cortex and an inner medullary region, which ultimately regresses, leaving behind a compressed nest of vestigial tubules and Leydig cells in the hilar region known as the rete ovarii. By 20 weeks of gestation, the ovary achieves mature compartmentalization, consisting of an active cortex containing follicles exhibiting early stages of maturation and atresia, and a developing stroma. Within the cortex, primordial follicles are separated from the somatic cells by a surrounding basement membrane. In some primordial follicles, the pregranulosa cells become cuboidal and proliferate, the ooycte enlarges and produces a zona pellucida (an extracellular glycoprotein matrix deposited between the ooycte and the granulosa cells), and a surrounding layer of thecal cells develops. The remainder stay quiescent until sometime later.
The molecular events that regulate primordial follicle formation and that stimulate or inhibit the initiation of follicular development are understood poorly but appear to involve a variety of factors, all locally produced and regulated, including members of the transforming growth factor β (TGF-β) superfamily of proteins and another family of trophic factors called neurotrophins. Activins, inhibins, antimüllerian hormone (AMH) and bone morphogenetic proteins (BMPs) are members of the TGF-β family of proteins. Activins promote and inhibins retard primordial follicle development, and their relative local concentrations in the fetal ovary during the time of follicle assembly may determine the size of the ovarian follicular pool.69 AMH appears to be an important inhibitor of primordial follicle growth, and BMPs exert the opposite effect.69 Neurotrophins and their receptors are essential for the differentiation and survival of various neuronal populations in the central and peripheral nervous systems, but their presence in the developing ovary suggests they also play a role in ovarian development. Four mammalian neurotrophins have been identified, including nerve growth factor (NGF), brain-derived neurotropic factor (BDNF), neurotrophin-3 (NT-3), and neurotrophin 4/5 (NT-4/5), all of which exert their actions via binding to high-affinity trans-membrane tyrosine kinase receptors encoded by members of the trk proto-oncogene family (NGF to TrkA, BDNF and NT-4/5 to TrkB, and NT-3 to TrkC).70 Observations in NGF- and TrkA-null mice indicate that NGF stimulates the proliferation of ovarian mesenchymal cells during the early stages of follicular assembly and promotes differentiation and synthesis of FSH receptors in granulosa cells. Similar experiments with TrkB-null mice suggest that TrkB signaling is required for oocyte survival after follicular assembly and for preantral follicular development.70 The specific signaling mechanisms that mediate the effects of activins, inhibins, BMPs and neurotrophins remain to be established.
Other paracrine factors mediate a bi-directional communication between oocytes and their surrounding granulosa cells. Oocytes are linked to their investment of granulosa cells via gap junctions which allow passage of small molecules such as ions (e.g., calcium), metabolites (e.g., pyruvate, nucleic acids, inositol), amino acids (e.g., L-alanine), cholesterol, and intracellular signaling molecules (e.g., cyclic adenosine monophophate, cAMP) between granulosa cells and oocytes. In mice, targeted deletions of gap junction proteins (known as connexins), disrupt follicular and oocyte development.68 Oocytes are unable to use glucose as an energy source to support meiotic maturation, cannot transport certain amino acids, and lack both the enzymes necessary for cholesterol synthesis and the receptors for its uptake from carrier-borne sources. Consequently, they are dependent on adjacent granulosa cells to metabolize glucose into a usable energy substrate, such as pyruvate, for transport of essential amino acids, such as L-alanine, and for synthesis and transfer of cholesterol.71 To meet their needs, oocytes stimulate glycolysis, amino acid transport, and cholesterol synthesis in granulosa cells via paracrine and juxtacrine signals that promote expression of transcripts involved in these metabolic processes, at least in some species.71 Candidate signaling molecules include closely related members of the TGF-β family, growth differentiation factor 9 (GDF9) and BMP15; both are expressed robustly in oocytes and appear crucial for normal ovarian follicle development in mammalian species.72
Genital Duct Differentiation and Development
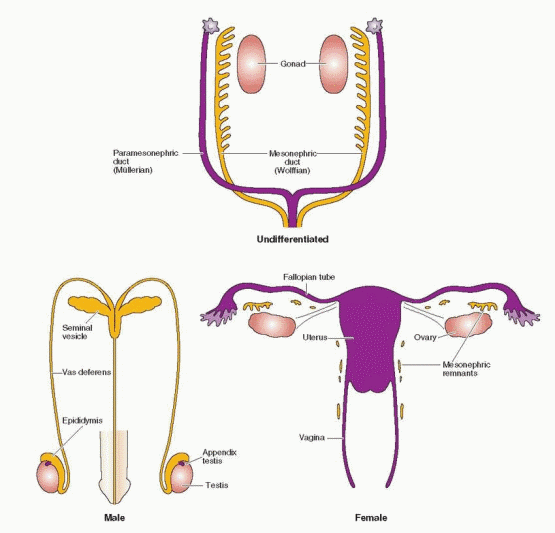
Caspar Wolff described the mesonephros in 1759 in his doctoral dissertation, at the age of 26.73 The paired structures were named wolffian bodies by the 19th century embryologist, Rathke, in recognition of Wolff’s initial discovery and description. Johannes Müller, a German physiologist, described the embyrology of the genitalia in 1830. The paramesonephric ducts received his name, not because of his original contributions, but in recognition of his ability to synthesize the existing literature into a coherent concept.
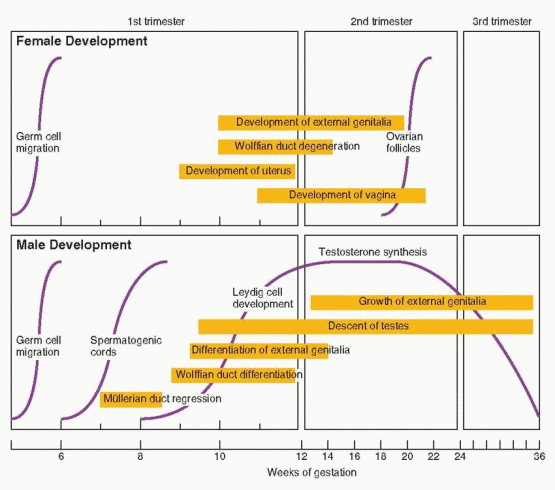
The mesonephric (wolffian) and paramesonephric (müllerian) ducts are discrete primordia that coexist in all embryos during the ambisexual period of development (up to 8 weeks). Thereafter, one duct system persists, giving rise to specialized ducts and glands, and the other regresses, leaving behind only nonfunctional vestiges. The wolffian duct develops first, differentiates into the epididymis, vas deferens, and seminal vesicles in males, and regresses in females. The müllerian duct develops later, even after the beginning of sex determination, differentiates into the fallopian tubes, uterus, and upper portion of the vagina in females, and regresses in males.
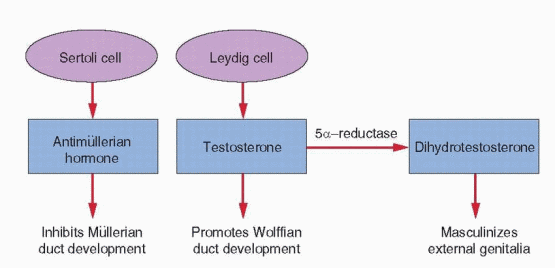
The hormonal control of genital duct differentiation and development was established by the classic experiments of Alfred Jost.74 His landmark studies demonstrated that hormones produced by the testis direct the sexual differentiation of both the internal and external genitalia in the male. Whereas testosterone stabilizes and promotes development of the wolffian ducts, AMH directs the regression of the müllerian system. In females, the wolffian ducts regress, in the absence of testosterone, and the müllerian ducts develop fully, in the absence of AMH. Although not yet clearly defined, our knowledge of the molecular mechanisms involved is growing steadily.
Mesonephric (Wolffian) Duct Development
Testosterone is secreted by the fetal testes soon after Leydig cell formation (at 8 weeks gestation) and rises rapidly to peak concentrations at 15-18 weeks. Fetal testosterone stimulates development of the wolffian duct system, from which the epididymis, vas deferens, and the seminal vesicles derive. Testosterone levels in the male fetus correlate with Leydig cell development, overall gonadal weight, 3β-hydroxysteroid dehydrogenase activity, and chorionic gonadotropin (hCG) concentrations. As maternal hCG levels decline, beginning at approximately 20 weeks gestation, Leydig cell testosterone secretion comes under the control of fetal pituitary luteinizing hormone (LH). In the absence of LH, as in males with anencephaly and other forms of congenital hypopituitarism, Leydig cells all but disappear and the internal and external genitalia do not develop fully.75
Testosterone can reach the developing wolffian duct system via the systemic fetal circulation, but the paracrine actions of testosterone produced in nearby Leydig cells are more important for the stabilization and differentiation of the wolffian duct. High local concentrations of testosterone stimulate the ipsilateral wolffian duct to differentiate into the epididymis, vas deferens, and seminal vesicle. Duct system differentiation proceeds, therefore, according to the nature of the adjacent gonad. High concentrations of testosterone are required because the duct does not have the ability to convert testosterone to dihydrotestosterone (DHT).76 In rodents, wolffian development can be induced in female embryos by treatment with exogenous androgens, but only to a limited extent,77 because exogenous androgen treatment cannot achieve and maintain the high local concentrations required to induce duct differentiation. For the same reason, the wolffian ducts do not develop in female fetuses exposed to excess endogenous adrenal androgens, as in
classical congenital adrenal hyperplasia, or to excess maternally-derived androgens, as occurs in women with pregnancy luteoma. Testosterone acts via binding to androgen receptors in the wolffian duct, which are detectable in both males and females, but androgen production in females does not approach the levels required to promote wolffian duct differentiation.77
classical congenital adrenal hyperplasia, or to excess maternally-derived androgens, as occurs in women with pregnancy luteoma. Testosterone acts via binding to androgen receptors in the wolffian duct, which are detectable in both males and females, but androgen production in females does not approach the levels required to promote wolffian duct differentiation.77
The paired wolffian ducts arise within the urogenital ridge during embryogenesis, running its length and terminating in the cloaca. The ducts form by a rearrangement of mesenchymal cells rather than by cell proliferation.78 The regulatory signals involved have not been established, but evidence from studies in mice having targeted deletions of candidate genes has implicated a number of transcription factors, including Pax2, Lim1, and Emx2. All are expressed in mesenchymal condensations before duct formation and respond to opposing signals from adjacent mesoderm and overlying ectoderm, which appear to restrict their expression to the specific area in the mesoderm from which the ducts arise.78 Along the axis of the forming wolffian ducts, a series of smaller tubules develop. The most anterior tubules fuse with the wolffian duct to become the precursors of the efferent ducts, ultimately connecting the testis to the epididymis; the more posterior or caudal tubules regress. In the human, parallel efferent ductules form multiple connections with the head (caput) of the epididymis.
Gradually, the straight wolffian ducts elongate and coil as a result of epithelial cell proliferation, stimulated by testosterone transported from the testes via the lumen of the duct, as well as by growth factors (e.g., epidermal growth factor, EGF; basic fibroblast growth factor, bFGF), which also are found in high concentrations in luminal fluid.79, 80 The structure of the developing epididymis becomes increasingly complex. Elongation and three-dimensional coiling begin at the end nearest the testis (the caput), and progress distally, except at the most caudal end of the duct, which remains straight and ultimately gives rise to the vas deferens. The factors that stimulate or control coiling of the duct are uncertain but may involve a combination of regional signals from the surrounding mesenchyme, focal “hot spots” of epithelial cell growth, and physical space limitations.78 Region-specific expression of homeobox (HOX) genes, which are transcriptional regulators of patterning, appears important for the differentiation of the duct into its morphologically and functionally distinct segments (caput, corpus, and caudal regions). For example, Hoxa10 and Hoxa11 appear to act distally to define the boundary between the epididymis and vas deferens.81 Others HOX genes appear to direct differentiation of the seminal vesicle (derived from the posterior wolffian duct) and the prostate (derived from the urogenital sinus).82 Evidence suggests that HOX genes may act by controlling the expression of other morphogenic factors such as inhibin beta A, which is expressed most highly in the greatly coiled caput region and to a progressively lesser extent in the mesenchyme surrounding more distal regions of the duct.83 Growth factors in the testicular fluid also appear to play an important role in cellular differentiation along the length of the epididymis.84
The extraordinary length of the epididymis—approximately 6 meters in the human— reflects its functional importance. As sperm leave the testis, they are functionally immature, having neither full motility nor the ability to recognize and fertilize an oocyte. They mature and acquire those functions as they pass through the epididymis, undergoing both biochemical and physical changes in a changing luminal environment regulated by a region-specific epididymal epithelium. The vas deferens is distinguished from the epididymis by its structure and by its function. It originates at the caudal end of the epididymis, where sperm are stored, and ends in the ejaculatory duct, which joins with the urethra. The vas deferens is surrounded by layers of smooth muscle that contract in response to sympathetic nerve stimulation, moving sperm through the vas deferens, into the ejaculatory duct (formed by the union of the vas with the duct of the seminal vesicle), and into the urethra.
Paramesonephric (Müllerian) Duct Development and Regression
The müllerian ducts begin by invagination of the coelomic epithelium, which progresses until reaching the wolffian ducts, then elongate, by cellular proliferation, along the length of the wolffian ducts until reaching and fusing with the urogenital sinus.85 The wolffian ducts make no direct contribution to the müllerian ducts, but are essential for normal müllerian development, serving as a guide or migrational template.86 If the wolffian ducts do not form, müllerian duct development also fails. Consequently, abnormalities in the renal system are highly associated with abnormalities in development of the fallopian tubes, uterus, and upper vagina.
Müllerian duct development can be separated into three phases, each controlled by different genes, as demonstrated by careful analyses of mutant mice. Selection of the cells in the coelomic epithelium that will become the müllerian ducts is controlled by Lim1, which encodes a protein also involved in formation of the wolffian ducts.87 Expression of Wnt4 and other genes in the Wnt family (Wnt7a, Wnt9b) appears necessary for epithelial invagination.85 Pax2 is required for duct elongation88 and, together with Pax8, also for differentiation of the duct into a uterus and vagina.89 Directly or indirectly, müllerian duct development also involves other genes such as those encoding retinoic acid receptors; mice having targeted deletions of the retinoic acid receptors fail to develop müllerian ducts or to differentiate specific portions of the duct.90
 |
AMH is a member of the TGF-β superfamily family of growth and differentiation factors that includes inhibin and activin.91, 92 The gene encoding AMH is located on the short arm of chromosome 19 (19p13.3). Like other members of the TGF-β superfamily, AMH signaling is mediated via a heterodimeric receptor consisting of a type I and a type II serine/threonine kinase receptor; the type II part of the receptor mediates ligand specificity and the type I receptor activates a downstream signaling cascade. The specific type II receptor that binds AMH, called AMHR2, has been isolated in several mammalian species; in the human, the gene encoding AMHR2 is located on chromosome 12 (12q13). Three different type I receptors have been linked to AMH signaling—ALK2, ALK3, and ALK6; ALK2 and ALK3 appear particularly important, because decreased expression or deletion of either disrupts müllerian duct regression.85 AMH gene expression is induced by SOX9 in Sertoli cells soon after testicular differentiation and results in the ipsilateral regression of the müllerian ducts by 8 weeks of gestation, before the emergence of testosterone and stimulation of the wolffian ducts.93 Inactivating mutations of AMH or AMHR2 result in persistent müllerian ducts in males.94
The process of müllerian duct regression involves a number of genes, but studies in mice indicate that Wt1 and Wnt7a play key roles. AMH signaling induces coelomic epithelial cells expressing Wt1, Amhr2, and Alk3 to migrate and surround the müllerian duct, transforming to mesenchymal cells in the process.95, 96 Wnt7a expression in the müllerian duct mesoepithelium promotes secretion of a signaling molecule (Wnt7a) that activates Amhr2 in the neighboring mesenchymal cells via Wt1, which binds and activates the Amhr2 promoter.85 At the same time, β-catenin gene expression increases in the mesenchymal cells surrounding the duct, and accumulation of b-catenin is accompanied by increased apoptosis in the müllerian ductal epithelium.95, 96 Whether Wnt-dependent β-catenin activity is required to induce Amhr2 expression or functions downstream of AMH signaling, or both, is not yet clear. Regardless, the process of müllerian duct regression appears to involve both apoptosis and the transition of ductal epithelial cells to mesenchymal cells.85 The matrix metalloproteinase MMP2 also plays a role, by mediating destruction of the extracellular matrix; available evidence indicates that MMP2 activity also is AMH-dependent, although the mechanism involved has not been established.97
 |
Development of the External Genitalia
In the bipotential state, which persists until 9 weeks of gestation, the external genitalia consist of a genital tubercle, a urogenital sinus, and lateral labioscrotal folds or swellings. Unlike the internal genitalia where both duct systems initially coexist, the external genitalia are neutral primordia able to develop into either male or female structures, depending on gonadal steroid hormone signals.
In the male, the Leydig cells of the fetal testis begin to secrete testosterone at 8-9 weeks of gestation and masculinization of the external genitalia begins one week later, at approximately 10 weeks. The genital tubercle grows, forming the penis, the edges of the urogenital sinus fuse to form the penile urethra, and the labioscrotal folds fuse to form a scrotum. The process typically is completed by 12 to 14 weeks of gestation. Thereafter, the principal change is in the growth and length of the penis. Complete development of the male external genitalia and differentiation of the prostate requires the conversion of testosterone to dihydrotestosterone (DHT), via the action of the intracellular enzyme 5α-reductase. The genital tubercle and the labioscrotal swellings are highly sensitive to DHT, being rich in both androgen receptors and 5α-reductase activity.
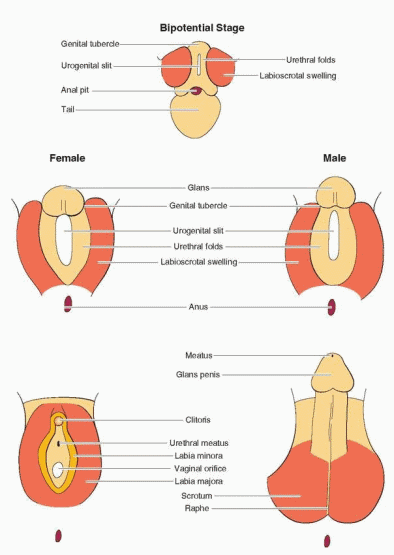 |
In the female, and in males with defects in androgen synthesis or action, the external genital primordia do not masculinize. The genital tubercle remains small and becomes the clitoris, the margins of the urogenital sinus remain separate and form the labia minora, the labioscrotal folds form the labia majora, and the urogenital sinus develops into the lower vagina and urethra.
In females, abnormal androgen exposure between 9 and 14 weeks of gestation results in varying degrees of masculinization, such as clitoral hypertrophy and labial fusion. In males, the external genitalia will not masculinize completely if androgen action is deficient during the same critical time interval, yielding a small phallus, hypospadias, or scrotal defects. In both sexes, because the external genitalia share a common origin, genital ambiguity results from abnormalities in androgen action—in females from too much, and in males from too little.
Sexual Differentiation of the Central Nervous System
Experimental evidence from studies in rodents and nonhuman primates suggests strongly that the fetal hormonal environment directs sexual differentiation of not only the genitalia, but also the central nervous system (CNS). Treatment with testosterone during early development increases reproductive and other behaviors more common in males and decreases behaviors more common in females. These observations suggest that testosterone and its metabolites play a role in brain development and neuronal organization.98, 99
Most of our knowledge about the early influence of testosterone on the brain and behavior in humans derives from clinical disorders associated with abnormal hormone production in early life, such as congenital adrenal hyperplasia (CAH). In male fetuses with classical CAH, sexual development progresses normally, but in female fetuses, testosterone is markedly elevated and causes masculinization of the external genitalia (clitoral enlargement and labial fusion). Studies in girls with classical CAH indicate that increased prenatal androgen exposure also affects their brains and behavior. Compared to unrelated age- and sex-matched controls or to unaffected female relatives of similar age, their toy preferences (vehicles, weapons) and play behaviors (rough, active play) are more typical of boys than of girls, to an extent that correlates with the severity of their disorder.4, 100, 101 Girls with classical CAH also display more physical aggression and greater spatial abilities.102,103 and 104 Although less well studied, there also is evidence to suggest that antenatal androgen exposure may influence sexual orientation. Whereas most females with classical CAH are heterosexual, as a group they are more likely to exhibit a bisexual or homosexual orientation; the effect is more pronounced in women with the severe salt-wasting form of CAH than in those with the milder, simple virilizing CAH.105 Other studies observing a significant linear relationship between childhood behaviors and maternal serum or amniotic fluid testosterone concentrations during pregnancy suggest that even normal variations in prenatal androgen exposure may influence behavior, in both males and females.106, 107
Presumably, the behavioral consequences of variations in prenatal androgen exposure reflect changes in neuronal development and organization. In rodents, an area of the anterior hypothalamic/preoptic region, called the sexually dimorphic nucleus of the preoptic area, is substantially larger in males than in females and treatment with androgens increases its size in females.98 Whereas no comparable, specific, sexually dimorphic region has been identified in the human brain, there is some evidence from studies in females with classical CAH using functional MRI to suggest that prenatal androgen exposure may
“masculinize” certain regions of the brain such as the amygdala, which is involved with regulation of emotion and aggression.108 Variations in fetal hormonal programming may contribute, therefore, to the spectrum of psychosexual behavior observed in humans. In addition, gender role is influenced heavily by the sex of rearing and by social interactions based upon genital appearance and secondary sexual characteristics.
“masculinize” certain regions of the brain such as the amygdala, which is involved with regulation of emotion and aggression.108 Variations in fetal hormonal programming may contribute, therefore, to the spectrum of psychosexual behavior observed in humans. In addition, gender role is influenced heavily by the sex of rearing and by social interactions based upon genital appearance and secondary sexual characteristics.
 |
Disorders of Sexual Development
Disorders of sexual development (DSD) are congenital conditions characterized by atypical development of chromosomal, gonadal, or phenotypic sex. Traditionally, they have been classified according to gonadal sex. A true hermaphrodite has both ovarian and testicular tissue. A male pseudohermaphrodite has testes, but a female genital phenotype, and a female pseudohermaphrodite has ovaries, but masculine genital characteristics. However, recent advances in molecular genetic diagnosis and increasing awareness of ethical issues and patient advocacy concerns suggested the need to re-examine the traditional classification scheme and to retire gender-based terms that many now consider pejorative.
Ideally, a classification system must be flexible, to allow incorporation of new information, logical, to maintain a consistent structure, reflect genetic cause when that is known, and accommodate the spectrum of phenotypic variation. The classification and nomenclature used here, organized by chromosomal composition and causation, conforms with recommendations arising from a 2006 consensus conference involving experts in pediatric endocrinology and other specialties involved in the management of patients with disorders of sexual development.109
46,XX Disorders of Sexual Development
Disorders of gonadal (ovarian) development
Ovotesticular disorder of sexual development (true hermaphroditism)
Testicular disorder of sexual development (46,XX male sex reversal)
Gonadal dysgenesis
Androgen excess—Fetal origin (congenital adrenal hyperplasia)
21-Hydroxylase (P450c21) deficiency
11β-Hydroxylase (P450c11β) deficiency
3β-Hydroxysteroid dehydrogenase deficiency
Androgen excess—Fetoplacental origin
Aromatase (P450arom) deficiency
P450 oxidoreductase deficiency
Androgen excess—Maternal origin (gestational hyperandrogenism)
Drug ingestion
Excess androgen production
Pregnancy luteoma
Theca-lutein cysts
Other disorders of genital development
Cloacal extrophy
Müllerian agenesis (Mayer-Rokitansky-Küster-Hauser syndrome)
Müllerian, renal, and cervicothoracic somite dysplasia (MURCS association)
46,XY Disorders of Sexual Development
Disorders of gonadal (testicular) development
Complete gonadal dysgenesis (Swyer syndrome)
Partial gonadal dysgenesis
Testicular regression syndrome
Ovotesticular disorder of sexual development
Disorders of androgen synthesis
Steroid 5α-reductase deficiency
17α-Hydroxylase (P450c17) deficiency
3β-Hydroxysteroid dehydrogenase deficiency
17β-Hydroxysteroid dehydrogenase deficiency
P450 oxidoreductase deficiency
Steroid acute regulatory (StAR) protein deficiency
Disorders of androgen action
Complete androgen insensitivity syndrome
Incomplete (partial) androgen insensitivity syndromes
LH receptor defects
Leydig cell hypoplasia
Disorders of antimüllerian hormone (AMH) and its receptor
Hernia uterine inguinale syndrome
Sex Chromosome Disorders of Sexual Development
45,X (Turner syndrome and variants)
47,XXY (Klinefelter syndrome and variants)
45,X/46,XY (mixed gonadal dysgenesis, ovotesticular disorder of sexual development)
46,XX/46,XY (chimerism, ovotesticular disorder of sexual development)
46,XX Disorders of Sexual Development
Disorders of sexual development in chromosomal females can result from abnormalities in gonadal development, but most are caused by androgen excess, which may be of fetal, fetoplacental, or maternal origin. Excess fetal androgen production results from steroidogenic enzyme deficiencies causing congenital adrenal hyperplasia. Androgen excess of fetoplacental origin results from enzyme deficiencies involving both the fetal adrenal and the placenta. Maternal androgen excess can result from the ingestion of drugs having androgenic properties and from disorders causing gestational hyperandrogenism.
Disorders of Gonadal (Ovarian) Development
Rarely, 46,XX disorders of sexual development (DSD) can result from abnormalities of gonadal development, which include ovotesticular DSD (true hermaphroditism), testicular DSD (46,XX sex reversal), and gonadal dysgenesis.
Ovotesticular Disorder of Sexual Development (True Hermaphroditism)
Ovotesticular DSD previously was called true hermaphroditism.109 Hermaphroditus, the Greek god with bisexual attributes, was the child of Hermes—the god of athletics, secrets, and occult philosophy—and Aphrodite, the goddess of love. The bisexual theme was immortalized in Greek and Roman sculptures depicting a woman with male external genitalia. Pliny (23-79 A.D.) was the first to apply the term hermaphrodite to humans, offering a description in his massive work, Historia Naturalis.
Ovotesticular DSD is a rare condition characterized by mixed ovarian and testicular tissue, which may include bilateral ovotestes or an ovotestis and a contralateral ovary or testis. The disorder is described here because the majority of patients have a 46,XX karyotype. However, because 7% of patients with ovotesticular DSD have a 46,XY karyotype and 10-40% exhibit chromosomal mosaicism,110 the disorder also must be listed among the causes of 46,XY- and Sex Chromosome Disorders of Sexual Development.
Whereas gonads containing testicular tissue are observed most frequently on the right, normal ovaries are observed most often on the left.110 Usually, both müllerian and wolffian internal genital structures are present and, as could be predicted, internal genital structures correspond with the adjacent gonad. Whereas most have a vagina, the uterus can be normal and functional, hypoplastic, vestigial, or altogether absent.110, 111 External genital development reflects the level of androgen production and exposure and the phenotype can range widely, from ambiguous genitalia to isolated hypospadias. Most are virilized sufficiently to allow male sex assignment, but three-fourths develop gynecomastia and half menstruate after puberty.
The genetics and pathophysiology of ovotesticular DSD are not well established. Mechanisms that might explain the testicular development include the translocation of testisdetermining genes from the Y to the X chromosome or an autosome, and autosomal dominant mutations that promote testis development in the absence of a Y chromosome.112
In one individual, the condition has been associated with an inactivating mutation in the RSPO1 gene,113 which is located on chromosome 1p34.2-3
In one individual, the condition has been associated with an inactivating mutation in the RSPO1 gene,113 which is located on chromosome 1p34.2-3
Testicular Disorder of Sexual Development (46,XX Sex Reversal)
Testicular DSD is a rare “sex reversal” syndrome in which the chromosomal sex (46,XX) is not consistent with the gonadal sex (testes). The disorder was first described by de la Chapelle in 1964,114 and can be divided into two types, SRY-positive and SRY-negative. Approximately 90% of cases result from abnormal recombination between the distal portions of the short arms of the X and Y chromosomes and transfer of SRY from the Y to the X chromosome during male meiosis; in 10% of cases, SRY cannot be detected.115 In most SRY-negative patients, the mechanism causing testis development cannot be determined.115,116,117 and 118
Although some patients with SRY-positive testicular DSD have ambiguous genitalia, which may result from preferential inactivation of the SRY-bearing X chromosome,119 the large majority are sterile males with normal genital development, a normal male hair pattern, and short stature. Consequently, unless they have cryptorchid testes, most are not recognized until after puberty, when they may present with hypogonadism, gynecomastia, and/or infertility.115 In contrast, SRY-negative XX males usually have ambiguous genitalia and often develop gynecomastia or fail to masculinize fully after puberty.115,116,117 and 118 Rarely, they may exhibit occult gonadal mosaicism for SRY.120 In some, the phenotype has been linked to a duplication of sequences on chromosome 17q, including the SOX9 gene, which acts downstream of SRY in the testis-determining pathway.32, 121 However, in most patients with SRY-negative testicular DSD, the cause remains unclear. In theory, XX male sex reversal might result from an inactivating mutation or deletion in genes encoding factors that inhibit testis development, but there is no direct evidence they are a cause of testicular DSD.116
Gonadal Dysgenesis
Some individuals with primary amenorrhea, hypergonadotropic hypogonadism, and
gonadal dysgenesis (streak gonads) have a normal 46,XX karyotype, providing indirect evidence that autosomal genes also play a critical role in ovarian differentiation. Affected women are normal in stature and, in most cases, have no apparent somatic anomalies. A wide variety of candidate genes have been identified, primarily via experiments involving murine knock-out models, including several that encode DNA and RNA binding proteins and transcription factors expressed during oogenesis.122
Androgen Excess—Fetal Origin (Congenital Adrenal Hyperplasia)
Virilizing CAH is a genetic disorder caused by enzyme defects in adrenal cortisol biosynthesis. More than 90% of cases result from a deficiency in the enzyme 21-hydroxylase.123,124 and 125 Deficiencies of 11β-hydroxylase and 3β-HSD are less common causes of CAH. In all, the pathophysiology relates primarily to decreased cortisol production, which stimulates a compensatory increase in pituitary adrenocorticotropic hormone (ACTH) secretion, causing adrenal hyperplasia; increased levels of steroid hormones proximal to the
enzyme block seek an alternative metabolic pathway, resulting in increased production of androgens.
enzyme block seek an alternative metabolic pathway, resulting in increased production of androgens.
In females, the classic forms of CAH (with and without salt-wasting) are characterized by genital ambiguity. Depending on the time, duration, and level of exposure, abnormally high androgen concentrations in utero result in varying degrees of clitoral enlargement and labial fusion and abnormalities of the urethra and vagina; generally, the urethra and vagina share a urogenital sinus that opens at the base of the clitoris. The fetal adrenal cortex does not achieve a significant level of function before 10 weeks gestation and, by that time, the vagina and urethra normally have become separated. However, between 10 and 12 weeks, rising androgen levels can promote progressive clitoral enlargement, labial fusion, and even partial closure of the urethra. At birth, the genital anatomy is similar to that in males with hypospadias and bilateral cryotochidism and can result in incorrect sex assignment. The effects of elevated adrenal androgen levels arising after 12-14 weeks of gestation are more limited. Female external genital development normally is not completed until approximately 20 weeks of gestation and the size of the clitoris depends more on the level than on the timing of excess androgen exposure. Development of the internal genitalia is normal in females with classical CAH because the excess androgen derives from the adrenals and the normal ovaries produce neither AMH nor significant amounts of androgen. Absent AMH and the high local androgen concentrations required to promote wolffian duct development, the fallopian tubes, uterus, and upper vagina develop normally.
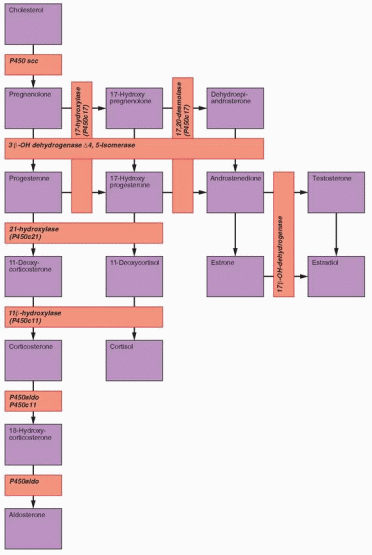
21-Hydroxylase (P450c21) Deficiency
The enzyme 21-hydroxylase (also designated P450c21 and CYP21A2) mediates the conversion of 17α-hydroxyprogesterone (17OHP) to 11-deoxycortisol (the immediate precursor of cortisol) and of progesterone to 11-deoxycorticosterone (an intermediate steroid in aldosterone synthesis). CAH due to 21-hydroxylase deficiency is the most frequent cause of sexual ambiguity and the most common endocrine cause of neonatal death. The more serious “salt-wasting” variety of classical 21-hydroxylase deficiency is characterized by severe deficiencies of both cortisol and aldosterone, resulting in salt-wasting and dehydration, in addition to virilization. In the less severe “simple virilizing” form of the disorder, elevated levels of ACTH are able to drive sufficient glucocorticoid and mineralocorticoid production to prevent circulatory collapse, but excess androgen production in utero results in masculinization of the external genitalia. The third and least severe “nonclassical” form of 21-hydroxylase deficiency generally does not become apparent until adolescence or early adulthood, when abnormally high androgen levels cause hirsutism and menstrual irregularities.
Data derived from neonatal screening programs for detection of classical CAH indicate that prevalence varies widely with ethnicity. Whereas the overall prevalence is approximatel 1 in 15,000 live births,126 prevalence ranges from 1 in 28,000 Chinese127 and between 1 in 5,000 and 1 in 23,000 Caucasians,128, 129 to as high as 1 in 280 Yupic Eskimos.130 In the United States, the prevalence of classical CAH is lower in African-Americans (1 in 42,000) than in Caucasians (1 in 15,500).131 Approximately two-thirds exhibit salt-wasting and onethird has the simple virilizing form of the disorder.
 |
Nonclassical 21-hydroxylase deficiency is one of the most common autosomal recessive diseases and, as in the classical form of the disorder, prevalence varies with ethnicity. Nonclassical 21-hydroxylase deficiency affects between 1 in 100 and 1 in 1,000 Caucasians,130,131 and 132 and may be even more common among those of Mediterranean, Hispanic, Slavic, and Eastern European Jewish descent.133 Most affected individuals are not identified in neonatal screening programs because their serum levels of 17OHP are not sufficiently elevated.134 Estimates of the carrier frequency (heterozygotes) for nonclassical 21-hydroxylase deficiency generally have ranged between 1 in 60 and 1 in 80 individuals,127, 130 but have been as high as 1 in 10 in a European population.135
All forms of CAH, including 21-hydroxylase deficiency, are transmitted as autosomal recessive disorders. Humans have 2 CYP21A genes; one is a nonfunctional pseudogene (CYP21A1, also designated CYP21P, encoding an inactive form of the enzyme), and the other is the active gene (CYP21A2). The 2 genes have greater than 90% homology and reside in the same region within the HLA histocompatibility complex on the short arm of chromosome 6 (6p21.3), which provides ample opportunity for recombination during meiosis.136,137,138 and 139 Most CYP21A2 mutations (approximately 75%) result from non-reciprocal gene conversions in which a segment of the CPY21A1 pseudogene is inserted into the active CYP21A2 gene, altering its sequence and resulting in point mutations that yield a defective enzyme.138,139,140,141 and 142 Approximately 20% of CYP21A2 mutations result from unequal cross-over exchanges between the two genes, yielding a larger fusion gene that produces an enzyme having reduced or no activity.128, 133, 142, 143 About 20 gene conversion mutations account for almost all of the affected alleles observed among various ethnic groups.141, 144,145,146,147,148,149,150 and 151 The remaining 5% of patients with CYP21A2 mutations have 1 or 2 of the more than 60 different point mutations that have been identified.141, 144,145 and 146
Women who carry a classic mutation are at risk for having a child with the severe form of the disorder. They may be asymptomatic, having one classic mutation and one normal allele, or exhibit the nonclassical form of CAH, having one classic mutation and a variant allele associated with mild enzyme deficiency (compound heterzygote). Compound heterozygotes having two variant alleles can exhibit the features of nonclassical CAH but are not at risk for having a child with classical CAH.
Although phenotype does not reliably predict genotype, the effect of a given mutation generally can be predicted by site-directed mutagenesis and expression, and by analysis of enzyme activity in vitro.132, 141, 147,148,149 and 150, 152,153,154,155,156,157,158 and 159
The salt-wasting form of classical 21-hydroxylase deficiency usually is associated with large gene deletions or a mutation that affects splicing and results in no enzyme activity.
Patients with the simple virilizing form of classical 21-hydroxylase deficiency most often have point mutations that result in low but detectable enzyme activity (e.g., 1-2% of normal) that supports adequate aldosterone and cortisol production.
Those with the nonclassical form of classical 21-hydroxylase deficiency usually are compound heterozygotes, having one classic mutation and one variant allele or two variant alleles; the phenotype of compound heterozygotes usually correlates with the less severe of the two mutations.144
Females with classical 21-hydroxylase deficiency (both salt-wasting and simple virilizing forms) present at birth with ambiguous genitalia (adrenogenital syndrome).162,163 and 164 Boys with salt-wasting CAH typically present as neonates or during early infancy with symptoms of adrenal insufficiency (failure to thrive, dehydration, hyponatremia, hyperkalemia), and those with simple virilizing CAH not identified by neonatal screening generally present as young children with early virilization. Females with the nonclassical “late-onset” form
of 21-hydroxlyase deficiency have normal external genitalia and present later, during childhood or early adolescence with precocious puberty, or as young adults with other signs of hyperandrogenism such as hirsutism.
of 21-hydroxlyase deficiency have normal external genitalia and present later, during childhood or early adolescence with precocious puberty, or as young adults with other signs of hyperandrogenism such as hirsutism.
As discussed earlier in this chapter in reference to the sexual differentiation of the CNS, females with classical CAH tend to exhibit greater interest in male-typical toys and play and more cross-gender and aggressive behavior than unaffected healthy women.4, 100,101,102,103,104 and 105 Studies of cognitive function in women with classical CAH have yielded inconsistent results. Whereas some have suggested that such women exhibit lower165, 166 or higher intelligence167 and differences in verbal learning and memory,168, 169 compared to unaffected women, others have found no evidence to indicate that prenatal androgen exposure has a consistent or predictable effect on cognition in women with CAH.170
Fertility in women with classical CAH is lower than in normal women,3, 105 primarily due to chronic anovulation relating to excess production of adrenal androgens and progestogens (progesterone, 17OHP) and disordered patterns of gonadotropin secretion;145 abnormalities of genital anatomy and psychological factors, such as delayed psychosexual development and decreased sexual activity, also contribute.171 In one study of quality of life in women with classical CAH, half reported that their disease adversely affected their sexual life and most were less than satisfied with their genital anatomy and function, regardless whether they had received reconstructive surgery; vaginal stenosis or narrowing were commonly observed.172 Women with classical CAH also had a later sexual debut and fewer pregnancies and children. Fertility rates correlate with the severity of the disorder and are significantly lower in women with salt-wasting than in those with the simple virilizing form of classical CAH.173 However, outcomes of pregnancies among women with classical CAH who conceive are normal except for an increased incidence of gestational diabetes.171 Children born to mothers with classical CAH have normal birthweight, no increased incidence of malformations, and exhibit normal intellectual and social development.171, 174 Although maternal serum androgen concentrations can increase significantly during pregnancy and should be monitored, the high capacity of placental aromatase activity effectively protects the female fetus from the masculinizing effects of maternal hyperandrogenism.174
Diagnosis of 21-hydroxylase deficiency is based on a high serum concentration of 17OHP, the primary substrate for the enzyme. In neonates with either salt-wasting or simple virilizing CAH, 17OHP levels typically are greater than 3,500 ng/dL;123, 175 levels in normal newborns generally are below 100 ng/dL.141 To distinguish 21-hydroxylase deficiency from other causes of CAH (11β-hydroxylase and 3βHSD deficiencies), serum concentrations of 11-deoxycortisol and 17α-hydroxypregnenolone also should be measured. When the diagnosis is suspected but uncertain, it can be confirmed by performing an ACTH stimulation test, obtaining blood samples before and 60 minutes after administering cosyntropin (synthetic ACTH 1-24; 1 μg/m2 or 0.25 mg);176 in affected infants, stimulated 17OHP levels typically exceed 10,000 ng/dL.162 Diagnosis also can be confirmed by genotyping, which can detect approximately 95% of mutations.177
In couples known to be at risk for having an affected child, (affected sibling, both partners carriers for a classic mutation), prenatal diagnosis is possible by genotyping amniocytes or, preferably, cells obtained by chorionic villus sampling (CVS).133, 146 Early prenatal diagnosis offers the option for intervention, before the most critical period of fetal genital differentiation, in efforts to avoid severe masculinization of the external genitalia in affected female fetuses.
Neonatal screening programs measure 17OHP in blood samples dried on filter paper, comparing results to established reference values that vary with weight and gestational age.178, 179 Antenatal corticosteroid treatment can decrease 17OHP levels and increase the risk for
a false negative result, particularly when administered repeatedly180; screening can be repeated at 1-2 weeks of age, with careful monitoring in the interim, or genotyping can be performed on the dried blood sample.181
a false negative result, particularly when administered repeatedly180; screening can be repeated at 1-2 weeks of age, with careful monitoring in the interim, or genotyping can be performed on the dried blood sample.181
In the late-onset nonclassical form of 21-hydroxylase deficiency, serum 17OHP concentrations often are only slightly elevated, especially late in the day, and the serum dehydroepiandrosterone sulfate (DHEAS) concentration usually is normal. In children, morning values greater than 82 ng/dL suggest the diagnosis, which can be confirmed by performing an ACTH stimulation test. In adult women, morning values less than 200 ng/dL (obtained during the early follicular phase of the cycle) exclude the diagnosis, levels over 800 ng/dL are virtually diagnostic, and intermediate results require additional evaluation with an ACTH stimulation test; in most patients with nonclassical 21-hydroxylase deficiency, the stimulated 17OHP level will exceed 1,500 ng/dL.133, 175, 182 A 21-hydroxylase deficiency can be distinguished from 11β-hydroxylase and 3βHSD deficiencies by also measuring 11-deoxycortisol and 17α-hydroxypregnenolone, but the distinction in patients with late-onset CAH has little or no clinical relevance and generally is unnecessary.
11β-Hydroxylase (P450c11) Deficiency
The enzyme 11β-hydroxylase (also designated P450c11 and CYP11B1) mediates the conversion of 11-deoxycortisol to cortisol and of 11-deoxycorticosterone to corticosterone (an intermediate steroid in aldosterone synthesis). The clinical features of 11β-hydroxylase deficiency result from the excess production of adrenal androgens and the mineralocorticoid action of 11-deoxycorticosterone; 11-deoxycortisol has no significant biological activity.
Although 11β-hydroxylase deficiency is the second most common cause of CAH, it accounts for only about 5-8% of adrenal steroid enzyme defects.162, 163, 183 Like 21-hydroxylase deficiency, 11β-hydroxylase deficiency has severe salt-wasting and simple virilizing forms, and a milder late-onset form. In females, 11β-hydroxylase deficiency can result in virilization of the external genitalia, but also may present later, in children with sexual precocity or in adolescent or young women with hirsutism and menstrual irregularity.184,185 and 186 In most affected individuals, the disorder has unique clinical features that help to distinguish it from 21-hydroxylase deficiency. Whereas both 21-hydroxylase deficiency and 11β-hydroxylase deficiency may result in salt-wasting, approximately two-thirds of patients with 11β-hydroxylase deficiency exhibit hypertension due to an increased production of mineralocorticoids.183, 187,188 and 189 Hypokalemia also may be observed and plasma rennin activity often is low. These effects generally have been attributed to excess production of 11-deoxycorticosterone, which has significant mineralocorticoid activity, although blood pressure and serum 11-deoxycorticosterone concentrations do not correlate closely.184, 190 The explanation for the wide variation in the clinical manifestations of 11β-hydroxylase deficiency is not clear.
The overall incidence of 11β-hydroxylase deficiency is approximately 1 in 100,000 live births, but like 21-hyroxylase deficiency, incidence varies with ethnicity. In Israel, the incidence of 11β-hydroxylase deficiency is as high as 1 in 5,000 births among Jews of Moroccan ancestry.191 The enzyme deficiency is an autosomal recessive disorder caused by mutations in the CYP11B1 gene, which is located on the long arm of chromosome 8 (8q21-q22). The known mutations include missense mutations that result in production of an inactive enzyme,159, 192,193 and 194 frameshift and nonsense mutations that prevent enzyme synthesis,195,196 and 197 and others resulting from unequal recombination between the CYP11B1 and CYP11B2 genes.198, 199 The CYP11B2 gene is located in the same region on chromosome 8 and encodes an enzyme having both 11β-hydroxylase and 18-hydroxylase
(also designated P450c18 or P450aldo) activity, mediating the conversion of corticosterone to 18- hydroxycorticosterone and, subsequently, aldosterone. There are no specific correlations between genotype and phenotype in patients with 11β-hydroxylase deficiency.200 Although the late-onset form of 11β-hydroxylase deficiency may be caused by mutations yielding an enzyme with reduced but still significant activity, none has yet been identified.
(also designated P450c18 or P450aldo) activity, mediating the conversion of corticosterone to 18- hydroxycorticosterone and, subsequently, aldosterone. There are no specific correlations between genotype and phenotype in patients with 11β-hydroxylase deficiency.200 Although the late-onset form of 11β-hydroxylase deficiency may be caused by mutations yielding an enzyme with reduced but still significant activity, none has yet been identified.
Diagnosis of 11β-hydroxylase deficiency is based on demonstrating high serum concentrations of 11-deoxycortisol and 11-deoxycorticosterone, as well as testosterone; both basal and ACTH-stimulated levels generally are elevated in affected neonates.183, 201, 202 In adolescents and young adults, basal 11-deoxycortisol and 11-deoxycorticosterone levels may be normal and ACTH stimulation often is required to make the diagnosis; results must be compared to established age and sex-specific normal values.
3β-Hydroxysteroid Dehydrogenase Deficiency
The enzyme 3β-hydroxysteroid dehydrogenase/Δ5-Δ4 isomerase (3β-HSD) catalyzes the oxidation and isomerization of Δ5-3β-hydroxysteroid precursors into Δ4-ketosteroids, an essential step in the formation of all classes of steroid hormones (glucocorticoids, mineralocorticoids, progestogens, androgens and estrogens). There are two 3β-HSD isoenzymes, designated type I and type II. The type I 3β-HSD gene (HSD3B1) mediates 3β-HSD activity in the placenta and peripheral tissues (skin, breasts, prostate) and the type II 3β-HSD gene (HSD3B2) is active in the adrenal, ovary and testis. Deficiency of type II 3β-HSD causes an uncommon form of CAH, accounting for less than 5% of cases.203 The type I isoenzyme is normal in patients with 3β-HSD deficiency. Consequently, serum concentrations of Δ4 steroids, such as 17OHP and androstendione, can be normal or even sometimes modestly elevated in affected patients. Serum levels of the substrates for the type I enzyme (pregnenolone, 17α-hydroxypregnenolone, DHEA) are increased due to the defect in the type II enzyme in the adrenals and gonads.
The clinical presentation of patients with 3β-HSD deficiency varies significantly, but can be divided into salt-wasting and non-salt-wasting forms. The salt-wasting form has been associated with nonsense mutations introducing stop codons,204 frameshift mutations,204,205 and 206 and a variety of point mutations in the HSD3B2 gene.207,208,209,210 and 211 Those with the non-salt-wasting form have had missense mutations causing single amino acid substitutions that dramatically decrease the enzyme’s affinity for substrates or cofactors.211,212,213 and 214
The external genitalia of females with 3b-HSD deficiency can be mildly virilized, presumably because DHEA levels are high and some is converted to androstenedione and, subsequently, to testosterone in the periphery. Whereas the salt-wasting form of classical 3β-HSD deficiency (analogous to those of 21-hydroxylase and 11β-hydroxylase deficiencies) usually is diagnosed during the first few months of life, the non-salt-wasting form of the disorder generally presents later. In females, because the external genitalia often are normal at birth, diagnosis of the non-salt-wasting form of 3β-HSD deficiency typically is delayed, presenting in childhood with premature puberarche, or in young women with signs of hyperandrogenism.203
Although basal levels of Δ5-3β-hydroxy steroids (pregnenolone, 17α-hydroxypreg-nenolone, DHEA and DHEAS) generally are elevated in affected individuals, an increased ratio of Δ5/Δ4 steroids is a better indication of a possible 3b-HSD deficiency. The most reliable diagnostic criterion is the serum 17a-hydroxypregnenolone concentration after ACTH stimulation. Proposed threshold values are based on observations in patients with documented mutations (neonates, ≥ 12,600 ng/dL; Tanner stage I children = 5,490 ng/dL, children with premature pubarche, = 9,790 ng/dL; adults = 9,620 ng/dL). Some
have argued that many women with a clinical diagnosis of polycystic ovary syndrome actually may have a late-onset form of 3β-HSD deficiency that may be as or more common than the late-onset form of 21-hydroxylase deficiency.215 An exaggerated 17α-hydroxypregnenolone response to ACTH stimulation is relatively common in women with hyperandrogenism, but levels rarely approach those observed in women with proven mutations, suggesting that the response likely reflects only adrenal hyperactivity and not an enzyme deficiency.216 Furthermore, molecular studies have only rarely identified any mutations in HSD3B2 in patients suspected of having a mild form of 3β-HSD deficiency.217,218 and 219
have argued that many women with a clinical diagnosis of polycystic ovary syndrome actually may have a late-onset form of 3β-HSD deficiency that may be as or more common than the late-onset form of 21-hydroxylase deficiency.215 An exaggerated 17α-hydroxypregnenolone response to ACTH stimulation is relatively common in women with hyperandrogenism, but levels rarely approach those observed in women with proven mutations, suggesting that the response likely reflects only adrenal hyperactivity and not an enzyme deficiency.216 Furthermore, molecular studies have only rarely identified any mutations in HSD3B2 in patients suspected of having a mild form of 3β-HSD deficiency.217,218 and 219
Treatment of Congenital Adrenal Hyperplasia
Treatment for classical forms of CAH is aimed at providing sufficient amounts of the deficient hormone, cortisol, to reduce excessive ACTH secretion and to prevent the consequences of excessive androgen production. In mothers at risk for having an affected child, treatment can reduce or prevent masculinization of a female fetus. In neonates with classical CAH, treatment can be life-saving and prevents further virilization. In children, treatment permits normal growth and sexual maturation. In adults with classical or nonclassical CAH, treatment helps in the management of hirsutism, menstrual abnormalities, and infertility.
Preimplantation Genetic Diagnosis in Couples at Risk for Having an Affected Child
Polymerase chain reaction (PCR)-based genotyping has greatly improved genetic counseling of families with CAH. In couples at risk for conceiving an affected child, the technology also can be applied in preimplantation genetic diagnosis (PGD) to detect affected embryos resulting from in vitro fertilization (IVF).220, 221 Typically, a single cell is removed from each embryo reaching the 6-8 cell stage on the third day after oocyte retrieval and fertilization by intracytoplasmic sperm injection (ICSI), with transfer of an unaffected embryo(s) 2 days later at the blastocyst stage. Although PGD requires IVF that otherwise would be unnecessary in fertile couples, some may prefer this option over others based on early prenatal diagnosis, as described below.
Prenatal Treatment of Mothers at Risk for Having an Affected Child
Prenatal maternal treatment with dexamethasone (up to 1.5 mg daily in divided doses) can greatly decrease or prevent fetal female genital virilization.222 Dexamethasone is not metabolized by the placenta and crosses effectively into the fetal circulation. For maximum effectiveness, treatment should begin at 4 to 5 weeks of gestation, and not later than 9 weeks.142, 222,223,224 and 225 Prenatal maternal treatment poses some potential risks for the fetus, such as postnatal failure to thrive and psychomotor developmental delay, and also can have significant maternal side effects, including severe abdominal striae, hyperglycemia, hypertension, gastrointestinal symptoms, and emotional lability.225, 226
Given that only one in eight fetuses will benefit from maternal treatment (one in four affected, half of which will be males), the best approach involves early prenatal diagnosis by CVS with rapid sex determination (fluorescence in situ hybridization for the X and Y chromosomes, or karyotype) and genotyping, continuing or beginning treatment only in those mothers having an affected female fetus. However, because even short-term prenatal treatment with dexamethasone may adversely affect postnatal physical, cognitive, and emotional development, careful pre-treatment counseling, monitoring, and long-term follow-up is required and best provided in a research setting.227, 228
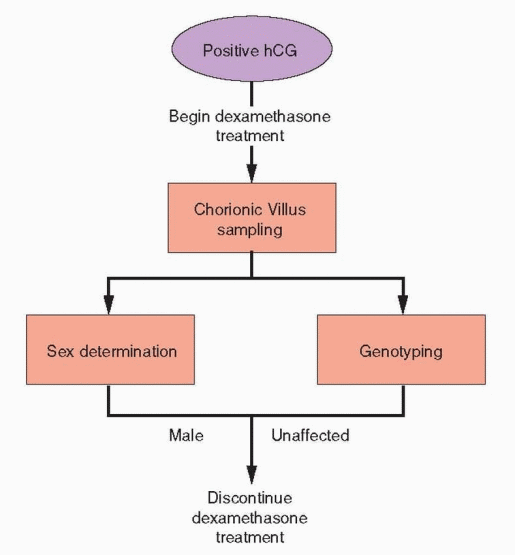 |
Neonatal Treatment
Newborn infants with classical CAH may be identified by prenatal diagnosis or neonatal screening, or because they have genital ambiguity (females), or an adrenal crisis (males). Infants who exhibit signs of adrenal crisis (hypotension, hyponatremia, hyperkalemia, hypoglycemia, vomiting and diarrhea, weight loss, anorexia) require urgent medical treatment, focusing first on administration of fluids (10-20 mL/kg 0.9% saline) and correction of any significant hypoglycemia (2-4 mg/kg 10% dextrose); hyperkalemia should be corrected by administering glucose and insulin, if necessary. After a blood sample is obtained for measurement of steroid hormones (17OHP primarily), a stress dose of hydrocortisone should be administered (50-100 mg/m2 intravenously, typically 25 mg), followed by 50-100 mg/m2 daily in divided doses (every four hours). Additional stress doses of hydrocortisone are administered until the infant is stable and feeding normally. Immediate mineralocorticoid replacement is not necessary but will be required if a diagnosis of salt-wasting CAH is confirmed. Initially, doses of fludrocortisone up to 0.3 mg daily and sodium chloride supplementation (1-3 g daily; 17-51 mEq daily) are required.
In infants having a positive neonatal screening test for CAH, the diagnosis should be confirmed with a second blood sample for measurement of 17OHP and electrolytes. While awaiting the results, electrolytes should be monitored closely if the infant is not treated empirically with glucocorticoids and mineralocorticoids. Again, the urgent need is to identify infants with salt-wasting CAH before they develop adrenal crisis, which can occur anytime within the first few days or weeks after birth without treatment.141, 222
Treatment in Children
Ideally, the medical, surgical, and psychological management of children with CAH should be guided by a multidisciplinary team, including pediatric endocrinologists, surgeons, urologists, geneticists, and psychologists.222
Children with classical or symptomatic nonclassical 21-hydroxylase deficiency require treatment with glucocorticoids.141, 222, 229 The goal of treatment is to promote normal growth and development by providing sufficient hormone to minimize adrenal sex steroid production while avoiding the consequences of glucocorticoid excess. Usually, that can be achieved by treatment with hydrocortisone (cortisol) in a dose of 12-18 mg/m2daily,133, 222, 229 which still exceeds normal daily cortisol secretion in children and adolescents (6-9 mg/m2/day).230,231 and 232 Whereas long-acting glucocorticoids (e.g., prednisone, dexamethasone) also can be used, their longer duration of action and greater potency also increase the risk of over-treatment, which can adversely affect growth before closure of the epiphyses.142, 233, 234 Normal growth has been observed in some studies of children treated with prednisone (approximately 1 mg/m2/day)235 or dexamethasone (approximately 0.27 mg/m2/day),236 but hydrocortisone remains the treatment of choice during childhood.222, 229
Mineralocorticoid treatment with fludrocortisone is required for children having classical 21-hydroxylase deficiency, regardless whether they have the salt-wasting or simple virilizing form of the disorder. The goal of treatment is to maintain normal serum sodium and potassium concentrations while avoiding the consequences of over-treatment or undertreatment. Excessive mineralocorticoid treatment can cause hypertension, hypokalemia, and may impair growth.237 Inadequate treatment can result in poor growth because it increases the glucocorticoid requirement,237, 238 and may increase adrenal androgen production, because chronic volume depletion causes increased production of renin and angiotensin II, which can stimulate steroidogenesis.239 In children, fludrocortisone is administered in a dose ranging between 0.05 and 0.2 mg daily.222 Salt supplementation can be discontinued as the child begins to eat table food, but may be needed during hot weather or strenuous exercise.
The effectiveness of treatment generally should be monitored approximately every 3 months in infants and every 4-12 months in children,222 by measuring the serum concentrations of 17OHP, androstenedione, plasma rennin activity, growth velocity, and skeletal maturation, comparing results to normative data for age and sexual maturation.133 Ideally, serum hormone measurements should be obtained in the morning when results will reflect peak concentrations.141, 222, 229 Serum 17OHP levels generally should be maintained in a range between 400 and 1200 ng/dL, but care must be taken to avoid undertreatment of hyperandrogenism and the consequences of iatrogenic hypercortisolism.240 Plasma rennin activity should be kept within the normal range for age by adjusting treatment with fludrocortisone and salt supplementation, before adjusting the level of glucocorticoid treatment. When necessary, a brief 7-10 day course of treatment with dexamethasone can effectively suppress high androstenedione levels that may result from poor compliance. Bone age and growth rate should be monitored every 6 months, with the goal of avoiding a decrease in growth and advanced bone age.241, 242 Patients with classical CAH have an increased risk for developing central precocious puberty due to poor control of adrenal androgen production; in those who do, treatment with a long-acting GnRH agonist may be needed.243
Illness can precipitate adrenal crisis in children with classical CAH unless they receive adequate glucocorticoid treatment. Signs and symptoms suggesting the possibility include hypotension, electrolyte imbalance (hyponatremia, hyperkalemia, hypoglycemia), and vomiting and diarrhea that sometimes can be accompanied by abdominal pain, fever, loss of appetite, and weight loss. In children with mild illness, the maintenance dose of glucocorticoid generally should be increased by 2- to 3-fold. When illness is associated with diarrhea or vomiting and reduced oral intake, intravenous glucocorticoids, saline, and glucose may be required. In children with severe illness or who require major surgery, intravenous hydrocortisone should be administered in a dose appropriate for age; for those 12 years of age or older, a one-time dose of 100 mg should be administered, followed by
100 mg/day. During recovery, stress doses of hydrocortisone can be gradually decreased, by approximately 50% per day.222
100 mg/day. During recovery, stress doses of hydrocortisone can be gradually decreased, by approximately 50% per day.222
Children with classical CAH are at increased risk for early puberty and short stature because high levels of sex steroids promote premature epiphyseal closure. In treated patients with classical CAH, adult height usually is lower than in reference populations, by an average of approximately 10 cm, independent of the level of control of adrenal androgen concentrations, which suggests that treatment with exogenous glucocorticoids also suppresses growth.244, 245 The effectiveness of treatment during the first 2 years of life and during puberty appears to have the most important influence on final height.246,247 and 248 Treatment with growth hormone and a long-acting GnRH agonist can help to maximize growth and adult height.249, 250 Obesity is a common complication of glucocorticoid treatment in children with classical CAH; body mass index correlates with the dose of prescribed medication.251 In obese children, the incidence of hypertension also is increased.252
The surgical management of the genital abnormalities in virilized female children with classical CAH is quite complicated. Traditionally, surgery has been performed in the first few years of life, when the child is still too young to remember the procedure, and to avoid any psychological problems associated with having abnormal external genitalia. However, the wisdom and outcomes achieved with early surgery recently have been challenged and many now advocate delaying unnecessary surgery until the child is older and can participate in the decision.109 The controversy is discussed in a later section of this chapter devoted to the management of ambiguous genitalia. If clitoroplasty is performed, the clitoral recession procedure, conserving the glans and its innervation, should be employed. It is important to know that women who undergo clitoroplasty and even total clitoral amputation generally do not have an impaired erotic response or decreased capacity for orgasm. When necessary, vaginal reconstruction is best postponed until after puberty when mature compliance is possible. In patients with severe classical CAH, bilateral adrenalectomy offers the potential advantage of preventing adrenal hyperandrogenism, but also increases the risk for developing adrenal crisis.253,254 and 255
Treatment in Adults
For older adolescent and adult women with classical CAH, the goal of treatment is to lower and maintain serum concentrations of adrenal precursors (17OHP) and androgens to the upper limits for normal women. After epiphyseal closure is complete, treatment with longacting glucocorticoids (e.g., dexamethasone, prednisone) generally is preferred. When administered at bedtime in a dose ranging between 0.25 and 0.75 mg, ACTH is effectively suppressed for most or all of the following day. Bedtime treatment effectively inhibits the peak of ACTH secretion, which occurs between 2:00 A.M. and 10:00 A.M.233 To avoid the risks of osteoporosis and developing Cushing’s syndrome, dosage must be adjusted to the needs of the individual patient. Alternative treatment regimens include prednisone (median dose 7 mg/day; range 4-10 mg/day) or single or divided doses of hydrocortisone (median 30 mg/day; range 15-40 mg/day).256 Supplemental doses of glucocorticoid, generally involving a 2-3 fold increase in the usual daily dose, are indicated during times of stress such as febrile illness, surgery, and trauma; normal exercise does not require stress doses of glucocorticoids.229
As in children with classical CAH, mineralocorticoid treatment in adults is provided with fludrocortisone, in the dose required to maintain normal serum sodium and potassium concentrations and plasma rennin activity, usually ranging between 0.1 and
0.2 mg/day. When mineralocorticoid treatment is optimized, the dose of glucocorticoids can be minimized.237, 238 Inadequate treatment can result in chronic volume depletion that promotes excess production of renin and angiotensin II, which, in turn, can stimulate increased adrenal androgen synthesis.239 Patients with the simple virilizing form of classical CAH who exhibit increased plasma renin activity and aldosterone concentrations can benefit from mineralocorticoid treatment, which helps in controlling 17OHP levels.257, 258
0.2 mg/day. When mineralocorticoid treatment is optimized, the dose of glucocorticoids can be minimized.237, 238 Inadequate treatment can result in chronic volume depletion that promotes excess production of renin and angiotensin II, which, in turn, can stimulate increased adrenal androgen synthesis.239 Patients with the simple virilizing form of classical CAH who exhibit increased plasma renin activity and aldosterone concentrations can benefit from mineralocorticoid treatment, which helps in controlling 17OHP levels.257, 258
Treatment should be monitored by periodic measurements of bone density, and serum 17OHP, DHEAS, androstenedione, and testosterone concentrations, remaining alert to the development of signs or symptoms of Cushing’s syndrome. In those who require mineralocorticoid treatment, plasma renin activity should be monitored and maintained near the upper limit of normal.
Many women with classical CAH who underwent reconstructive surgery during childhood later require further reconstructive surgery during late adolescence or early adulthood, generally involving clitoroplasty and vaginoplasty. Approximately half of procedures performed during infancy will require later revision.133, 259
Psychological counseling, ideally beginning soon after the diagnosis is established, is an important part of the treatment of classical CAH. Although data are limited and conflicting, the incidence of adult psychiatric disorders may be increased in women with classical CAH.260, 261 Sexual relationships may develop somewhat later than usual and sexual function may not be completely normal, even in those having had reconstructive surgery.5
Treatment During Pregnancy
Although normal reproduction is possible with effective treatment, fertility generally is decreased in women with classical CAH, particularly in those with the salt-wasting variety of the disorder, due to chronic anovulation and, in some cases, to poor surgical results.3 In those who do conceive, serum concentrations of androstenedione, testosterone and 17OHP should be carefully monitored and the dosage of glucocorticoids increased as needed to maintain normal levels for gestational age. Treatment with long-acting glucocorticoids should be discontinued in favor of treatment with hydrocortisone, which is metabolized by the placenta and thereby avoids the risk of suppressing the fetal hypothalamic-pituitary-adrenal axis. In general, term pregnancies, delivery of healthy female infants with normal external genitalia, and normal growth and development in both girls and boys can be achieved.174, 262 Even when maternal androgen levels cannot be suppressed to normal, the high capacity of placental aromatase activity effectively protects the fetal female genitalia.174
The incidence of cesarean delivery is increased, primarily because of concerns that vaginal delivery may disrupt a previous surgical reconstruction of perineal anatomy. An android pelvis is no more common than usual, because the form and size of the adult pelvis are determined during the pubertal growth spurt. However, a small pelvis might result if bone age is advanced to age 13-14 before treatment started. The need for stress doses of glucocorticoids during labor and delivery is obvious and does not increase the risk for infection or poor wound healing.
Androgen Excess—Fetoplacental Origin
Two rare enzyme deficiencies associated with androgen excess—aromatase deficiency and P450 oxidoreductase deficiency—are distinct from those causing classical forms of CAH because they involve both the fetal adrenal and the placenta.
Aromatase (P450arom) Deficiency
The enzyme aromatase (also designated P450arom and CYP19A1) catalyzes the conversion of 19-carbon androgens (androstenedione, testosterone, 16α-hydroxy DHEA) to aromatic 18-carbon estrogens (estrone, estradiol, and estriol, respectively) and is encoded by the CYP19A1 gene, located on chromosome 15 (15p21.1). The enzyme is active in the gonads, the placenta, the brain, and in adipose; tissue-specific regulation is controlled, in part, by alternative tissue-specific promoters. Aromatase deficiency is a rare autosomal recessive disorder caused by mutations in the CYP19A1 gene. As a consequence, fetal androgens are not converted to estrogens in the placenta, resulting in female fetal virilization (due to the accumulation of fetal androgens), low maternal serum estrogen levels, and maternal hirsutism, which typically develops during the second half of pregnancy and regresses after delivery. Affected females classically present with ambiguous genitalia at birth and, at puberty, exhibit signs of hyperandrogenism, absent breast development, primary amenorrhea associated with hypergonadotropic hypogonadism, and multicystic ovaries.263,264,265 and 266 Aromatase mutations also can produce variable or nonclassic phenotypes characterized by varying degrees of breast development.267
P450 Oxidoreductase Deficiency
The classical forms of CAH all are caused by mutations in genes encoding steroidogenic enzymes, resulting in reduced or absent enzyme activity, and in clinical signs and symptoms caused by the accumulation of steroid precursors and/or decreased production of the principal steroid end product. Another newly described form of CAH results from a deficiency in the P450 oxidoreductase (POR) enzyme. Although not a steroidogenic enzyme per se, POR nonetheless affects several steroidogenic pathways and now is recognized as a cause of both 46,XX disorders of sexual development (female virilization) and 46,XY disorders of sexual development (incomplete male virilization), which is discussed below.268
First described in 2004,269 POR deficiency is perhaps the most complex form of CAH because it affects the activity of all of the P450 enzymes involved in steroidogenesis, to varying degrees, resulting in varying patterns of abnormal steroid hormone production and a spectrum of clinical manifestations, and has other “non-endocrine” effects on skeletal development and drug metabolism. POR is a flavoprotein associated with the endoplasmic reticulum and is encoded by the POR gene, located on chromosome 7 (7q11.2). POR serves as the electron donor in the activation of all microsomal P450 enzymes, including P450c21 (the adrenal 21-hydroxylase, CYP21A2), P450c17 (CYP17A1, which catalyzes both 17α-hydroxylase and 17,20-lyase activities), and P450arom (aromatase, CYP19A1, which mediates the conversion of androgens to estrogens). POR deficiency is an autosomal recessive disorder and more than 25 different POR mutations already have been identified, most being missense mutations in the central electron transfer domain of the protein.268
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


