Neoplasia
Robert J. Arceci
Howard J. Weinstein
Although neoplasia in infancy is quite rare, it presents important and unique biologic, diagnostic, and therapeutic problems. Many tumors in early life are composed of persistent embryonal or fetal tissues, suggesting a failure of proper maturation or cytodifferentiation during intrauterine or early postnatal life. The failure of proper maturation of fetal tissue may be sometimes difficult to distinguish from neoplasia. Additionally, an unexpectedly large number of neoplasms of early life are associated with growth disturbances and congenital anomalies. Spontaneous regression and cytodifferentiation also occur most frequently in tumors of early life. The unique physiology of the developing neonate provides the clinician with special problems in terms of therapeutic interventions and their long-term sequelae, including secondary malignancies.
▪ EPIDEMIOLOGY
From the data in the Third National Cancer Survey (1969 to 1971), Bader and Miller (1) reported that, in the United States, the annual incidence of malignant neoplasms in infants younger than 1 year of age was 183.4 per 1 million live births and within the first 28 days of life was 36.5 per 1 million live births. They further estimated that approximately 653 infants per year in the United States are diagnosed with cancer and that about 130 (20%) of these patients are neonates. In a later study from Denmark, the incidence of neonatal cancer was calculated to be in a similar range, with values of 1.88 to 2.98 cases per 100,000 births (2). Approximately one-half of the neonatal malignancies are noted on the first day of life. In contrast, the annual incidence of cancer in the United States for persons under the age of 20 years is approximately 15 per 100,000. There does not appear to be any gender bias.
When the incidence for all malignancies is compared with mortality as determined from death certificates, the incidence in patients younger than 1 year is 3.5 times greater than mortality, whereas the incidence in patients younger than 29 days old is 4.8 times greater than mortality. In contrast, in children up to 15 years of age, the incidence of malignancy is only about 1.3 to 1.8 times greater than mortality. There are also marked differences in incidence versus mortality when specific types of cancer are considered. For example, in neonates, the incidence of neuroblastoma is 10 times greater than the mortality, whereas the incidence of leukemia is only 1.8 times the mortality.
Further, the distribution of the types of malignancies found in infants younger than 1 year of age differs from that found in later childhood. For example, neuroblastoma is the most common malignancy in infants under 1 year of age and accounts for about 50% of malignancies in the neonatal period; it is followed by leukemia, renal tumors, sarcomas, central nervous system (CNS) tumors, and hepatic malignancy (3). However, when one considers the total spectrum of neoplastic disorders of infancy, teratoma is usually reported as the most frequently encountered neoplasm, followed by hemangiomas, lymphangiomas, and small nevi lesions. In children younger than 15 years of age, leukemia is the most common malignancy (about 30%), followed by CNS tumors, lymphoma, neuroblastoma, sarcoma, and renal tumors. Thus, the incidence and types of neoplastic disorders of infancy contrast greatly compared to later childhood and define the neonatal period as epidemiologically distinct in terms of these disorders.
▪ ORIGINS AND CAUSES OF NEONATAL CANCER
Developmental Growth Disturbances, Genetic Aberrations, and Cancer Pathogenesis
Naturally occurring DNA sequences homologous to transforming viral oncogenes exist in normal, untransformed cells of all metazoa. Such DNA sequences, termed cellular oncogenes, are used in normal cells to control growth, development, and differentiation in precise temporal and tissue-specific patterns. Because of their expression and critical role during normal development, inherited or acquired mutations affecting cellular oncogene expression and/or function can lead to a variety of developmental abnormalities and congenital defects, such as hemihypertrophy syndromes and hamartomas. Additionally, the persistent expression beyond birth of certain growth-related oncogenes may play a role in such proliferative states as the transient myeloproliferative disorder (TMD) associated with Down syndrome and stage IV-S neuroblastoma found in infants, both of which are characterized by subsequent, spontaneous regression.
Inherited syndromes usually occur as a result of chromosomal aneuploidy, deletions, translocations, increased fragility, or altered epigenetic imprinting. An example of aneuploidy is Down syndrome (trisomy 21), in which the frequency of acute leukemia is approximately 15 times the normal. Additionally, an increased incidence of solid tumors has been reported for persons with trisomies 8, 9, 13, and 18 (4). Deletion of part of the long arm of chromosome 13 is associated with psychomotor retardation, microcephaly, cardiac and skeletal defects, and the early development of retinoblastoma. The deletion of the short arm of chromosome 11 results in mental retardation, microcephaly, aniridia, ear and genital anomalies, and an increased incidence of Wilms tumor (WAGR syndrome). These syndromes provided support for the assignment of a retinoblastoma-associated locus to chromosome 13q14 and a Wilms tumor-associated locus to 11p13 followed by identification of the retinoblastoma (RB) and the Wilms tumor genes (WT1). These mutant genes are usually heterozygous in germline DNA and homozygous in tumors, thus resulting in the loss of heterozygosity characteristic of tumor suppressor genes. WAGR syndrome results from loss of several genes from the 11p13 region. Deletion of one copy of PAX 6 is responsible for aniridia, and loss of one WT1 allele results in genitourinary anomalies. Homozygosity at the Wilms tumor locus on chromosome 11 has also been found in embryonal rhabdomyosarcomas (RMS) and hepatoblastomas, suggesting a common pathogenesis for these embryonal tumors. The specific loss of constitutional heterozygosity and its relationship to oncogenesis has been confirmed in studies of transgenic mice that lack a functional tumor suppressor gene, p53. The inherited human counterpart, termed Li-Fraumeni syndrome in humans, is characterized by a higher incidence of embryopathy and an increased incidence of malignancies developing early in life.
A number of inherited syndromes, including Bloom syndrome, Fanconi anemia, ataxia-telangiectasia, xeroderma pigmentosum, and Werner syndrome, are characterized by developmental abnormalities and increased incidence of various types of cancer (5). Many of these syndromes are known to be caused by defects in genes encoding key proteins involved in DNA recombination and repair, such as in Fanconi anemia or Bloom syndrome or excision repair enzymes associated with xeroderma pigmentosum. It is of
note that when these defective genes are inherited through the germ line, patients show both developmental abnormalities and an increased incidence of cancer, thus linking genes leading to developmental defects to cancer predisposition.
note that when these defective genes are inherited through the germ line, patients show both developmental abnormalities and an increased incidence of cancer, thus linking genes leading to developmental defects to cancer predisposition.
Malformations and malformation syndromes without obvious cytogenetic abnormalities include hemihypertrophy and Beckwith-Wiedemann syndrome (BWS), which consists of mental retardation, gigantism, macroglossia, omphalocele, and organomegaly as well as being associated with the development of Wilms tumor, hepatoblastoma, and adrenocortical carcinoma. BWS, which occurs in approximately 1 in 13,000 births, is usually sporadic, although an autosomal dominant inheritance pattern with incomplete penetrance has also been proposed. Patients with BWS have an approximately 7.5% to 10% risk of developing a tumor.
Hamartomas are pathologically benign proliferations of cells in their normal anatomic location. Hamartomas in which malignant neoplasms arise include congenital melanotic nevi, which can progress to melanoma, and familial polyposis, which may evolve into colonic carcinoma. Examples of malignancies developing from persistent fetal rests include craniopharyngioma, which arises from tissue derived embryologically from the Rathke pouch, and neuroblastoma arising from persistent adrenal neuroblasts.
These predisposing conditions share at least one common element: an inherited or developmental disturbance of cellular growth and/or cell survival, which may be linked to the molecular pathways regulating these genetically determined cellular responses. The identification of these different classes of genes helps to define the molecular links between conditions of abnormal development (i.e., teratogenesis) and neoplastic transformation.
Prenatal Exposure to Maternal Genotoxins
There are relatively few reports and/or studies of outcome in infants born to mothers undergoing chemotherapy and/or radiation therapy for cancer. The risk of developmental problems increases with decreased gestational age at the time of maternal exposure. For example, a recommendation to terminate a pregnancy is commonly used when significant numbers and doses of anticancer drugs are used during the first trimester because of the increased risk of major birth defects and spontaneous abortions. Outcomes for infants whose mothers are treated during the second and third trimesters are significantly better, although there has been reported some risk of low birth weight, intrauterine growth restriction, and stillbirths (6,7).
Some prenatal drug exposures may lead to increased risk of cancer in offspring. For example, prenatal exposure to diethylstilbestrol has been closely linked to the development of clear cell adenocarcinoma of the vagina, Dilantin exposure with neuroblastoma, nitrosourea compounds with CNS tumors, and topoisomerase II inhibitors (epipodophyllotoxins, flavonoids, catechins, caffeine) with leukemia associated with mixed lineage leukemia (MLL) gene rearrangements. Significant use of alcohol and tobacco/marijuana and exposure to pesticides have been reported to be associated with an increased risk of congenital leukemia, although this association remains controversial (8). Radiation therapy or significant exposures to radiation through diagnostic testing such as computed tomography (CT) scanning are usually avoided whenever possible in pregnant mothers because of concerns of potential morbidity and cancer risk to the developing fetus.
Exposure to Maternal Malignancy
In addition to the susceptibility of the fetus to adverse effects of chemotherapy during pregnancy, there is also the possibility that the maternal cancer will metastasize to the placenta and fetus. Although many anecdotal reports have documented such involvement, it occurs only very rarely. The types of tumors transmitted from the mother to the placenta or fetus are quite varied, with melanoma most commonly cited. Although lymphoma and leukemia may involve the placenta, they have usually not been found to be transmitted to the fetus. The evaluation of infants born to mothers with cancer has not been clearly established, in part because of the rarity of such events. However, close follow-up is recommended during the first year of life, including physical exams, blood studies such as complete blood count (CBC) and liver function tests, and scans only when clinically indicated. The frequency of transfer of a maternal cancer to an infant appears to be higher when the infant suffers from immunodeficiency. Careful examination of the placenta is an important component of this evaluation.
▪ SPECIFIC TUMORS AFFECTING NEONATES
Tumors of Neuroepithelial Origin
Neuroectodermal cells of the neural tube differentiate to neuroblasts, which can then develop into several key cell lineages and subtypes, including nervous system tissue and melanocytes; free spongioblasts, which become either astrocytes or oligodendroglia cells; and ependymal spongioblasts, which become ependymal cells. Neoplasia may arise in any of these primitive neuroectodermal cellular compartments, giving rise to a group of morphologically similar tumors involving central and peripheral sites of the nervous system. Neonatal tumors originating from neuroectodermal cells include neuroblastoma, retinoblastoma, peripheral nerve tumors (i.e., neuroepithelioma), medulloblastoma, choroid plexus papilloma, ependymoblastoma, and melanotic neuroectodermal tumors. These tumors show varying degrees of cellular differentiation, have similar histologic features (e.g., small, primitive cells with rosettes or pseudorosettes), and tend to spread along cerebrospinal fluid pathways. The most common and clinically important are discussed in more detail.
Neuroblastoma
Neuroblastoma is the most common malignant tumor in neonates. It originates from neural crest cells that normally give rise to the adrenal medulla and sympathetic ganglia. Occurrence in siblings and other family members suggested that some cases are hereditary and led to the identification of germline mutations, particularly in ALK and PHOX2B, in familial neuroblastoma (9,10,11). In such cases, the tumors are usually diagnosed at an earlier age and often characterized by having multifocal primary tumors (12). An interesting syndrome has been reported in several women who delivered infants diagnosed as having neuroblastoma during the first few months of life. The mothers had sweating, pallor, headaches, palpitations, hypertension, and tingling in their hands and feet during the last trimester of pregnancy. These symptoms resolved after the birth of the affected infants, suggesting that the symptoms were caused by the fetal tumor catecholamines transmitted into the maternal circulation.
Although at least half of infants with neuroblastoma present with an abdominal mass from tumors arising in the adrenal medulla or retroperitoneal sympathetic ganglia, neuroblastoma may arise anywhere along the sympathetic nervous system and/or present with disseminated disease. In adrenal neuroblastoma, an abdominal sonogram or CT scan demonstrates displacement of the kidney without distortion of the calyceal system. The neoplasm also may originate in the posterior mediastinum, neck, or pelvis. Cervical sympathetic ganglion involvement may result in Horner syndrome; mediastinal tumors may cause respiratory distress; paravertebral tumors tend to grow through the intervertebral foramina and cause symptoms of spinal cord compression; and presacral neuroblastomas may mimic presacral teratomas. Neuroblastoma also has been detected prenatally by ultrasonography, showing a solid and sometimes cystic suprarenal mass. Two unusual presentations of neuroblastoma are intractable diarrhea secondary to release of
vasoactive intestinal peptide, and the paraneoplastic syndrome of opsoclonus, myoclonus, and truncal ataxia. The diarrhea secondary to vasoactive intestinal peptide abates after removal of the neuroblastoma. In contrast is the unpredictable improvement after the removal or treatment of neuroblastoma associated with opsoclonus-myoclonus.
vasoactive intestinal peptide, and the paraneoplastic syndrome of opsoclonus, myoclonus, and truncal ataxia. The diarrhea secondary to vasoactive intestinal peptide abates after removal of the neuroblastoma. In contrast is the unpredictable improvement after the removal or treatment of neuroblastoma associated with opsoclonus-myoclonus.
Metastatic lesions are common presenting findings of neuroblastoma in the neonatal period. The primary tumor often cannot be found in infants younger than 6 months of age. These infants present with bluish, subcutaneous nodules and extensive hepatomegaly. The liver may be studded with tumor nodules and be so large that it causes respiratory distress secondary to abdominal distention. Clumps of tumor cells often are found in the bone marrow aspirates. Metastases to bones, skull, and orbit, which present as periorbital ecchymoses, are, however, rare in the neonate. The unique metastatic pattern to liver, bone marrow, and skin in infants is classified as stage IV-S neuroblastoma (13).
The differential diagnosis for neuroblastoma is limited. The subcutaneous nodules appear similar to those found in congenital leukemia cutis and several congenital infections. The leukoerythroblastosis secondary to bone marrow metastases from neuroblastoma also is observed with congenital infection, severe hemolytic disease, and leukemia. More than 90% of children with neuroblastoma will have elevated urinary excretion of catecholamine metabolites, vanillylmandelic acid or homovanillic acid, or both. The diagnosis of neuroblastoma is made by biopsy of the primary tumor or metastatic lesions. The most histologically primitive lesion is neuroblastoma without differentiation and is composed of small, round cells with scant cytoplasm. The ganglioneuroma, its benign counterpart, is composed of large, mature ganglion cells. Ganglioneuroblastoma has components of both. In the absence of a tissue specimen, the findings of elevated urinary catecholamines and tumor pseudorosettes in a bone marrow specimen can be sufficient to make a definitive diagnosis.
The prognosis for children with neuroblastoma is in general inversely correlated to the age of the child at diagnosis and the extent of disease. Infants with stage IV-S frequently have spontaneous regression of disease and may undergone maturation into mature ganglioneuroma. The incidence of spontaneous regression of neuroblastoma may be more common than is clinically evident. Primitive sympathetic neuroblasts, which are derived from neural crest ectoderm, migrate in early embryonic life into the adrenal primordium, in which they arrange themselves in nodules before differentiation into adrenomedullary tissue. These nodules are present in all fetal adrenal glands at 14 to 18 weeks of gestation. Beckwith and Perrin (14) detected the presence of microscopic clusters of neuroblastoma cells (i.e., neuroblastoma in situ) in the adrenal glands in a number of autopsies from infants younger than age 3 months who had no clinical evidence of tumor. They estimated that neuroblastoma in situ occurs in 1 of 250 stillborn infants and infants younger than 3 months of age. However, clinically detectable neuroblastoma is noted in only 1 of 10,000 live births. Also pertinent to these observations are the results from neonatal catecholamine screening programs, which have demonstrated that this type of screening resulted in an increased incidence of early-stage neuroblastoma that, most likely, would not have presented as clinically detectable disease (15,16).
The extent of treatment for neuroblastoma depends on the stage of the disease and biologic factors such as histology, MYCN amplification, and DNA ploidy. An infant with stage IV-S disease should usually be observed for a period of weeks to months before treatment is initiated because of the reasonable likelihood of spontaneous regression. However, respiratory difficulties, blood vessel (usually vena cava) obstruction, and gastrointestinal compression secondary to rapid tumor expansion often require treatment. Single or multiagent chemotherapy (often carboplatin) is usually the first line of treatment (12). About 8% to 10% of patients with stage IV-S disease may have MYCN amplification and are then recommended to receive treatment according to high-risk regimens.
Complete surgical removal of neuroblastoma is usually accomplished in infants with stage I or II disease. Postoperative treatment is not generally indicated for these patients, and their long-term survival is excellent (12). Similarly, many infants with stage III disease in whom gross residual tumor remains after surgery may not require further therapy (17). Chemotherapy is used for infants with localized or stage IV disease in conjunction with immune/cytokine treatment and autologous transplantation (18). The active chemotherapeutic agents against neuroblastoma include alkylating compounds (e.g., cisplatin, temozolomide, cyclophosphamide, dacarbazine), vincristine, and doxorubicin. Outcomes for infants with intermediate-stage neuroblastoma have a greater than 90% overall survival, while those with MYCN amplification have an approximately 50% overall survival, suggesting that the biology plays a more significant role in prognosis than age alone (19).
Retinoblastoma
Retinoblastoma is a congenital malignant tumor arising from the nuclear layer of the retina and is the most common ocular tumor of childhood. The median age at presentation is 18 months and 14 months for bilateral cases, but a small percentage of infants are diagnosed during the first few months of life. Prenatal diagnosis by sonography of retinoblastoma has been observed. Approximately 10% of children with retinoblastoma have a family history of the disease while about 30% with bilateral or multifocal unilateral tumors have a negative family history (20). These two groups are capable of transmitting the disease to their offspring in an autosomal dominant fashion. This inheritance pattern is due to mutations in the retinoblastoma (RB) gene located on chromosome 13q14. Retinoblastoma has also been associated with chromosomal 13 deletion mosaicism). If there is a family history of retinoblastoma, an experienced ophthalmologist should examine the eyes of unaffected siblings regularly. Moll and associates have proposed such screening until age 4 years (21).
The most common initial signs of retinoblastoma include an abnormal white pupil (i.e., leukocoria), known as a cat’s-eye reflex and a squint or strabismus. Other potential diagnoses that can resemble retinoblastoma include granulomatosis uveitis, congenital defects, and severe retrolental fibroplasia. Once the diagnosis of retinoblastoma is suspected, both eyes should be examined with the infant under general anesthesia (22). A bone marrow aspiration and spinal tap for malignant cells are often performed for staging, although bone marrow examination has more recently been found to be less critical (23). A staging system for retinoblastoma is based on the size, location, number of tumors in each eye, and distant hematogenous metastases. Vitreous seeding, tumors extending anteriorly to the ora serrata, tumors invading over one-half of the retina, residual orbital disease, and optic nerve or distant metastases are all adverse prognostic features.
Brain Tumors and Other Neuroectodermal Tumors
Intracranial tumors presenting in the first year of life are uncommon. Brain tumors in children in this age group tend to be supratentorial, in contrast to those of older children, which are usually infratentorial. In infants, the most common presenting symptom is macrocrania, with a bulging fontanelle secondary to either hydrocephalus or tumor volume. Seizures, vomiting, failure to thrive, abnormal eye movement, and irritability also are frequent. The histologic diagnoses of the neuroectodermal tumors are similar to those of tumors in later childhood, with gliomas accounting for most.
Teratomas have been reported to represent the single most frequent intracranial tumors in neonates (26).
Teratomas have been reported to represent the single most frequent intracranial tumors in neonates (26).
Desmoplastic infantile gangliogliomas are rare, but massive, cystic tumors that usually occur supratentorially in the neonatal period (27). They present most commonly with signs of increased intracranial pressure, including seizures. Therapy has included surgery and chemotherapy, but without radiation therapy. For patients who have a complete surgical resection, additional therapy may not be required. Prognosis may be better than that observed with other tumors, such as high-grade astrocytoma. In contrast, pineoblastomas are malignant tumors with an extremely poor outcome, even when treated with surgery, chemotherapy, and radiation therapy. Part of the reason for the poor outcome in these infants may be the propensity of pineoblastoma to involve the leptomeninges and extraneural spread.
Treatment of infants with brain tumors historically included surgical removal or biopsy followed by radiotherapy. Operative mortality was high, and few infants survived for longer than 1 year. In these few, brain radiotherapy resulted in severe intellectual and psychomotor retardation (28). In attempts to avoid the adverse effects of radiotherapy on the developing brain, subsequent therapeutic approaches have used surgery followed by preradiation chemotherapy or intensive chemotherapy without radiation (29).
Atypical teratoid/rhabdoid tumors (AT/RT) of the CNS, although rare, are usually large and aggressive and have historically had very poor outcomes with the use of conventional therapies used in children with brain tumors. More aggressive, multidisciplinary approaches have started to show some improvement in patients with this type of brain tumor, although median survival is still estimated to be less than 2 years (30,31). These tumors all share the inactivation of the SNF5/INI1 gene, which encodes for a key developmental chromatin remodeling factor (32).
Primitive neuroectodermal tumors of peripheral nerve represent a group of soft tissue tumors known as neuroepitheliomas, medulloepitheliomas, and peripheral neuroblastomas (33). They are associated with major branches of peripheral nerves (i.e., tumor of the chest wall arising from intercostal nerve). These are extremely rare tumors, quite aggressive in their biologic behavior, with frequently occurring distant metastases, including to the CNS. Treatment approaches include wide excision, if possible, and chemotherapy modeled after either neuroblastoma or brain tumor protocols.
The melanotic neuroectodermal tumor of infancy has its origin in the neural crest population. Most of these tumors are diagnosed between 1 and 8 months of age; they commonly occur in the maxilla, although extremely rare cases have been reported in other sites such as the epididymis. They are considered a benign neoplasm, with a local recurrence rate of about 15%. These tumors originate from pluripotential neural crest cells that give rise to both melanoblasts and neuroblasts. The rate of malignant transformation for this tumor is reported to be approximately 5%. Recommended treatment is wide local excision.
▪ CONGENITAL LEUKEMIA
Leukemia in the newborn is extremely rare with approximately five cases reported per million live births (34). It has been customary to categorize leukemia as congenital when it is diagnosed within a few days after birth and as neonatal when it manifests itself during the first 4 to 6 weeks of life. The kinetics of leukemic cell growth and the estimated leukemic cell burden at the time of diagnosis make it reasonable to assume that clinically detectable leukemia during the first 4 weeks of life originated in utero (35,36). Molecular studies indicate a prenatal initiation of acute lymphoblastic leukemia (ALL) and some acute myeloid leukemias, in children diagnosed in infancy or even later in life.
Evidence for a genetic predisposition to acute leukemia is its occurrence in identical twins in which there is a very high concordance. If leukemia develops in one of a set of identical twins before 6 years of age, the risk of disease in the other twin is nearly 100%. Leukemia usually develops in the other twin within weeks to months of the first case. In some of these cases, intrauterine exchange of leukemic cells from one twin to the other has been strongly suggested by the identity of molecular and genetic changes observed postnatally in the leukemias from each twin (35,36). For fraternal twins and siblings, the risk of development of leukemia is two and four times higher than in the general population. Congenital leukemia has been associated with trisomy 9, trisomy 13, Turner syndrome, and Down syndrome and rearrangements of the MLL (KMT2) gene.
More than 95% of the childhood leukemias, including congenital leukemia, are classified as acute, because they are characterized by a predominance of immature lymphoid or myeloid precursors. The proportion of cases of ALL to acute myelogenous leukemia (AML) is approximately four to one in children, but this ratio is reversed in the congenital leukemias. Whereas lymphoblasts from most children with ALL express the common acute lymphoblastic leukemia antigen (CALLA or CD10) on the cell surface and express markers of pre-B-cell differentiation (e.g., cytoplasmic immunoglobulin or Ig gene rearrangement), the lymphoblasts of congenital and infant ALL appear earlier in B-cell lineage development, being CD10 negative. These infants have a higher incidence of CNS leukemia at diagnosis, a higher leukocyte count, increased frequency of hepatosplenomegaly, and a poorer prognosis than older children with ALL (37). A translocation of the long arms of chromosomes 4 and 11, t(4;11), involving the MLL gene at chromosome 11q23, is commonly observed in infant leukemia. This translocation is associated with greater than 80% of ALL in infancy and carries a poor prognosis (37). The most common subtype of AML in the neonate is acute monocytic leukemia, which accounts for only 20% of AML in older children, and this is also commonly associated with MLL rearrangements (37).
Cutaneous manifestations are the most frequent clinical findings noted at birth. In addition to petechiae and purpura, leukemic skin nodules (i.e., leukemia cutis) have been observed in approximately 50% of cases. These skin nodules may vary in size from a few millimeters to a few centimeters, are bluish to slate gray in color, may appear in all sites, and are palpated as firm tumors of the deep skin (Fig. 49.1). Neonatal leukemia cutis may undergo a spontaneous, temporary regression, but tends to recur in a more generalized form within a few weeks to months. Hepatosplenomegaly is common, but lymphadenopathy is not. Respiratory distress, secondary to leukostasis within the pulmonary vasculature, may complicate the clinical course. Other nonspecific symptoms of neonatal leukemia include lethargy, pallor, poor feeding, and umbilical, gastrointestinal, or genitourinary bleeding. The diagnosis of leukemia is confirmed by examination of a bone marrow aspirate obtained from the posterior iliac crest, although peripheral blood immunophenotyping and cytogenetic and molecular analyses are being increasingly used.
A variety of disorders in the newborn imitate leukemia. The newborn bone marrow response to infection, hypoxemia, or severe hemolysis commonly is a leukemoid reaction and an increase in circulating nucleated erythrocytes. These conditions can easily be confused with congenital leukemia.
Transient myeloproliferative syndrome occurs primarily in infants with Down syndrome. This syndrome, noted during the first few days of life, mimics AML. Peripheral leukocyte counts can range from 25,000 to several hundred thousand; bone marrow aspirates reveal 30% to 70% blasts. Hepatosplenomegaly and thrombocytopenia are also common findings. The hematologic status of these neonates usually returns to normal in 1 to 4 months, with only supportive therapy. Several of these children who subsequently died of cardiac or pulmonary disease years after the resolution of their transient myeloproliferative syndrome showed no evidence of leukemia at autopsy. This syndrome has been observed in neonates with
stigmata of Down syndrome and in phenotypically normal infants who have trisomy 21 mosaicism in their hematopoietic cells or skin fibroblasts. Molecular studies have identified the presence of truncated GATA1 mutations in essentially all cases of transient myeloproliferative syndrome and the acute megakaryoblastic leukemia associated with Down syndrome (38,39). GATA1, a key transcription factor that regulates megakaryopoiesis and erythropoiesis, has thus been etiologically linked in these two hematologic disorders and in the increased drug sensitivity of leukemic megakaryoblasts from children with Down syndrome. There have also been reports of spontaneous remissions of congenital leukemia, even in those children without Down syndrome, suggesting that some cases can be initially conservatively managed with observation and/or supportive care.
stigmata of Down syndrome and in phenotypically normal infants who have trisomy 21 mosaicism in their hematopoietic cells or skin fibroblasts. Molecular studies have identified the presence of truncated GATA1 mutations in essentially all cases of transient myeloproliferative syndrome and the acute megakaryoblastic leukemia associated with Down syndrome (38,39). GATA1, a key transcription factor that regulates megakaryopoiesis and erythropoiesis, has thus been etiologically linked in these two hematologic disorders and in the increased drug sensitivity of leukemic megakaryoblasts from children with Down syndrome. There have also been reports of spontaneous remissions of congenital leukemia, even in those children without Down syndrome, suggesting that some cases can be initially conservatively managed with observation and/or supportive care.
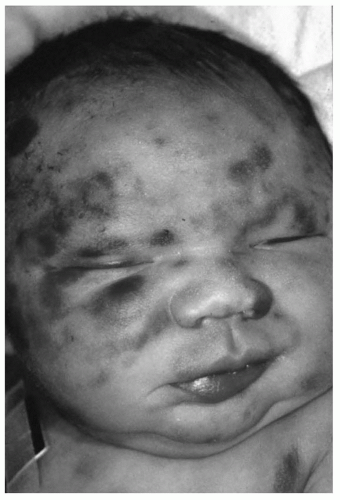 FIGURE 49.1 Congenital acute monocytic leukemia with skin nodules. |
Congenital ALL should be managed with systemic chemotherapy. Age has been an important prognostic variable in childhood ALL, with the most favorable prognosis for patients between 2 and 9 years of age. Prognosis for young infants with ALL remains poor (37). Intensive combination chemotherapy for neonates and infants with AML treatment regimens has significantly improved long-term disease-free survival (40,41).
▪ NEOPLASMS OF THE KIDNEY
Mesoblastic Nephroma
Most abdominal masses presenting in infancy are renal in origin, and most can be accounted for by cystic disease of the kidney and congenital malformations of the urinary tract leading to hydronephrosis. Although neoplasms of the kidney are rare in infancy, they do occur and have important prognostic implications, making it mandatory that they be included in the evaluation of abdominal masses.
The most common renal tumor found in infants is mesoblastic nephroma, which accounts for nearly 80% of renal tumors in the neonatal period. It also has been called fetal renal hamartoma, mesenchymal hamartoma of infancy, and leiomyomatous hamartoma. Mesoblastic nephroma commonly presents as an asymptomatic, enlarging abdominal mass during the first few months of life. It is not associated with congenital anomalies and has no race predilection. Of note is the more frequent occurrence of polyhydramnios and premature labor in women whose infants have mesoblastic nephroma. The differential diagnosis includes renal cystic disease, congenital malformations of the urinary tract resulting in hydronephrosis, and Wilms tumor. The chromosomal translocation resulting in a fusion protein, ETV6-NTRK3, has been reported in both mesoblastic nephroma and infantile fibrosarcoma (42).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


