Movement Disorders, Tics, and Tourette Syndrome
Diana Apetauerova
Movement disorders are defined as either a loss or poverty (akinesia) or slowness (brady-kinesia) of movement that is not associated with weakness or an excess of abnormal involuntary movements. Based on this, movement disorders are classified as either hypokinetic (Parkinsonism) or hyperkinetic (tremor, dystonia, chorea, tics, myoclonus). Movement disorders are generally caused by abnormalities in basal ganglia and their connections. The basal ganglia are that group of gray matter nuclei lying deep within the cerebral hemispheres (caudate, putamen, and pallidum), the diencephalon (subthalamic nucleus), and the mesencephalon (substantia nigra). The causes of many movement disorders remain unknown, in others various causes have been identified ranging from environmental toxins, genetic causes, medications, metabolic disorders, structural lesions, neurodegenerative causes, infectious, postinfectious causes, and autoimmune and psychogenic causes. More recent genetic, biochemical, and functional imagine advances have provided additional information about the pathophysiology and etiology of some movement disorders. Many diseases have now been localized to a specific gene (PD, dystonia, ataxia, paroxysmal dyskinesia, etc); several inherited movement disorders are due to expanded repeats of the trinucleotide cytosine-adenosine-quanosine (CAG) such as Huntington disease, some spinocerebellar ataxias (SCAs), and Dentatorubral and Pallidoluysian Atrophy (DRPLA).
APPROACH TO THE PATIENT
Movement disorders must be seen, because description of them by the parents or patient might be vague and will not lead to a proper diagnosis. Children also manifest a variety of intriguing physiologic and developmental abnormalities that await proper classification, and only the experienced movement disorders observer can distinguish those from movement disorders.
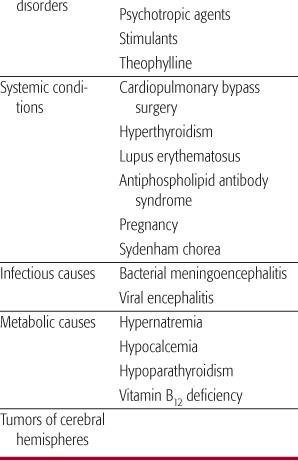
Many abnormal movement disorders are paroxysmal or at least intermittent; they can be induced by sleep, emotional upset, movement, or other triggering factors. The physician needs to ask what are triggering factors and what makes the movement better and worse, if there is fluctuation of symptoms during the day, and if there are any associated features (ie, loss of consciousness or awareness). Many paroxysmal movements in pediatric population are associated with epilepsy, and this has to be to exclude by appropriate investigation (see Chapter 557). Movement disorders may be classified as follows:
Hyperkinetic movement disorders
Chorea and athetosis
Dystonia
Tremor
Myoclonus
Tics and Tourette syndrome
Stereotypy
Hypokinetic movement disorders
Parkinsonism
HYPERKINETIC MOVEMENT DISORDERS
 CHOREA AND ATHETOSIS
CHOREA AND ATHETOSIS
• Chorea is a combination of fluid or jerky movement affecting any part of the body; chorea can resemble a dance (from Greek word), and movements are repetitive but not rhythmic or stereotyped. Patients with chorea appear restless.
• Athetosis is a slow, writhing movement of the limbs that may occur alone but is often associated with chorea (choreoathetosis).
• Ballismus is a high-amplitude, violent flinging of a limb from the shoulder or pelvis and is considered to be an extreme form of chorea.
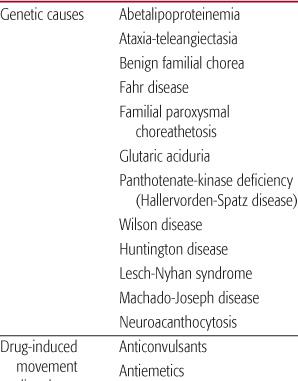
Common causes of chorea are listed in Table 566-1.
Cardiopulmonary Bypass Surgery
Chorea is seen in 1% to 10% of children with congenital heart disease following cardiopulmonary bypass surgery and profound hypothermia.1 The mechanism of basal ganglia injury is unknown. Choreoathetosis begins within 2 weeks postoperatively and may be associated with orofacial dyskinesia, hypotonia, affective changes, and pseudobulbar signs. Outcome varies; spontaneous resolution may occur, but significant morbidity and even mortality may result. MRI of the brain is normal. Treatment includes sedation in severe cases.
Drug-Induced Chorea
Drug-induced chorea is common after administration of dopamine antagonists and is usually dose related. Chorea usually presents in a form of tardive dyskinesia (TD). TD denotes chorea that occurs late (tardive) in the course of the use of medication or when medication is discontinued. This occurs commonly with chronic use of neuroleptics. The incidence of TD in children taking neuroleptics is estimated to be 1%.
TD often affects the region of the mouth and face and includes chewing-like movements, tongue protrusion, and lip smacking. TD can involve other parts of the body including the trunk and extremities. Symptoms occur from months to years after starting therapy. The condition should be suspected in any child who is chronically on neuroleptics and should be distinguished from tics and Tourette syndrome. TD should be treated by discontinuation of the offending medication or by switching to an atypical neuroleptic. Addition of benzodiazepines can be useful. 
 BENIGN HEREDITARY CHOREA
BENIGN HEREDITARY CHOREA
This is a rare disorder that presents with chorea in early childhood and is inherited in an autosomal dominant pattern.2 Chorea can be associated with delayed motor development, tremor, dysarthria, and hypotonia. Children with this disorder have a positive family history. The therapy recommended is low doses of neuroleptics.
Fahr Disease
Fahr disease is a combination of encephalopathy and progressive calcification of basal ganglia. It may be familial or sporadic and can be transmitted in an autosomal-dominant or autosomal-recessive pattern. Patients present with chorea and mental deterioration. Other clinical features can include ataxia, dysarhria, seizures, dwarfism, senile appearance, and retinitis pigmentosa. Patient’s symptoms progress into early disability and death. CT and MRI show calcification in the basal ganglia. There is no specific therapy for this disorder.
Hallervorden-Spatz Disease (Panthotenate-Kinase Deficiency)
Panthotenase-kinase deficiency is a rare hereditary condition of abnormal iron metabolism, transmitted in an autosomal-recessive pattern. Patients are affected at 2 to 10 years of age, and clinical manifestations include chorea, dystonia, rigidity, and dysarthria. Other less common features include retinitis pigmentosa, seizures, and mental retardation. Genetic testing is available to establish the diagnosis and consists of a defect in the gene PANK2 on the short arm of chromosome 20 encoding the enzyme Pantotenate Kinase 2.3 Neuroimaging studies often reveal a heterogeneous signal intensity in the basal ganglia, which constitutes a feature highly suggestive of this disorder (“eye-of-the-tiger”).4 Symptomatic therapy with levodopa might improve symptoms of rigidity; anticholinergics can improve dystonia. Symptoms usually progress, and death occurs in the early 30s.
Systemic Lupus Erythematosus (SLE)
Chorea associated with SLE is an uncommon early presentation of the disease. Clinical features are indistinguishable from Sydenham chorea. Patients might have additional neurological symptoms such as ataxia, psychosis, or seizures. The typical duration of the symptoms is about 12 weeks, but one fourth of the patients have a recurrence. The diagnosis is clear if the patient has history of LE; if chorea is the initial presentation, it is important to differentiate it from Sydenham chorea. Laboratory tests include elevation of sedimentation rate and antinuclear antibodies. Therapy includes high doses of corticosteroids.
Sydenham Chorea (SCH)
SCH is the most common cause of acquired chorea in children and occurs most commonly between ages 5 and 15 years. It is a cardinal feature of rheumatic fever and is sufficient alone to make a diagnosis. SCH usually occurs several weeks to months after untreated streptococcal infection (beta hemolytic streptococcus). The disorder may result from an abnormal immune reaction in which antibodies produced in response to the invading bacterium act against basal ganglia.5
Patients may also present with hypotonia and behavioral changes. The majority of patients will have gradual improvement of the symptoms within weeks; recurrence of chorea is common, occurring in about 20% of patients. Rheumatic valvular disease develops in about one third of untreated patients.
Diagnosis is made by the clinical presentation and cannot be confirmed by laboratory tests. Because the onset of symptoms is delayed for some time after infection, the antistreptolysin ASO titer can be normal or only slightly elevated. Brain MRI may show increased T2 signal in the putamen and globus pallidus, which resolves when chorea resolves.6 All children with SCH must be treated with penicillin in high doses for 10 days. Some experts suggest that patients with acute rheumatic fever should be prophylactically treated for 5 years up to age 18 (whichever comes first). A longer period of prophylaxis is recommended only for those who have significant rheumatic valvular heart disease (see Chapter 235).
Huntington Chorea
Huntington disease (HD) is an autosomal dominant neurodegenerative condition. The classic signs include chorea and dementia with personality changes. Although symptoms typically become evident during the fourth and fifth decades, the age of onset ranges from early childhood to late adulthood.
HD is transmitted as an autosomal dominant trait. The disorder occurs as the result of abnormally long CAG repeats of coded instructions within a gene on chromosome 4 (4p16.3).7 The progressive loss of nervous system function associated with HD results from loss of neurons in the basal ganglia and cerebral cortex. Juvenile HD is called Westphal variant and presents with rigidity, bradykinesia, dystonia, myoclonus, tremor, behavioral problems, and seizures.
 DYSTONIA
DYSTONIA
Dystonia is an involuntary muscle contracture caused by simultaneous contracture of agonist and antagonist muscle which causes twisting of various body parts (face, neck, limb, and trunk). Dystonia can produce writhing involuntary movements or can cause fixed abnormal posture. Dystonia can affect a single body part (focal dystonia), 2 or more contiguous body parts (segmental dystonia), the arm and leg on one side of the body (hemidystonia), or multiple body parts (generalized dystonia). Most dystonia in children begins focally and spreads over time to the rest of the body. A characteristic feature of dystonia is a presence of “sensory tricks” (gestes antagonists). Patients can temporarily suppress dystonic movements by those tricks. These tricks usually consist of touching the affected or adjacent body parts.
Classification of dystonia has been based upon etiology, age of onset, or body part affected. Historically, dystonia has been divided into primary (idiopathic) and secondary. In the future, classification may rely entirely on identification of genetic defects that either cause the disease or provide predisposition to the development of dystonia (eTables 566.1, 566.2, and 566.3  ).
).
The most common dystonias encountered in children are dopa-reponsive dystonia and idiopathic generalized torsion dystonia.
Dopa-Responsive Dystonia (Segawa Disease, DRD)
DRD is the most common cause of primary dystonia with onset in childhood. DRD presents with gait disturbance as a result of foot dystonia. The symptoms show diurnal variation with worsening later in the day and marked improvement in the morning. If dystonia remains untreated, parkinsonian symptoms may also develop later in the course of the illness. The age group most commonly affected is 1 to 12 years. Characteristically, DRD shows a dramatic response to low doses of L-dopa, without development of long-term treatment complications.
DRD is caused either by GTP cyclohydrolase deficiency (more common) or by tyrosine hydroxylase (TH) deficiency.8,9 GTP cyclohydrolase is the initial and rate-limiting enzyme involved in the biosynthesis of tetrahydrobiopterin (BH4), which is a cofactor for tyrosine hydroxylase, the rate-limiting enzyme in dopamine synthesis. The most common type of inheritance is autosomal dominant. Diagnosis is made by mutation analysis.
Idiopathic Torsion Dystonia (ITD, Dystonia Musculorum Deformans)
ITD is an autosomal dominant condition with incomplete penetrance. The cause of ITD is GAC deletion in the DYT-1 gene at 9q34.10 The product of the DYT-1 gene is called torsin A.11
Symptoms of ITD may appear early in life at about 9 years of age but may be delayed until about 45 years or later. Dystonia usually first occurs in the limbs as a focal dystonia, but over time spreads to become generalized. Trunk dystonia is a characteristic feature in younger patients. Patients do not have other neurological symptoms, and they do not develop mental deterioration.
Therapy usually considered is a trial of levodopa, but this medication is not effective in ITD. Trihexyphenidyl can be beneficial, as well as baclofen and diazepam. Deep brain stimulation surgery in the globus pallidus pars interna brought promising results in therapy of this debilitating disorder. 
 GLUTARIC ACIDEMIA TYPE I
GLUTARIC ACIDEMIA TYPE I
Glutaric acidemia type I presents with an acute encephalitis-like illness with somnolence, irritability, subsequent development of chorea and dystonia. Diagnosis and treatment are discussed in Chapter 140.
 TREMOR
TREMOR
Tremor is an involuntary oscillating movement with a fixed frequency. The most common cause of tremor in children is essential (familial) tremor. Essential tremor (ET) is transmitted in autosomal dominant trait.12 Patients with ET at young onset do not develop other neurological symptoms. ET can occur as early as 2 years of age. The most commonly involved areas of the body are the hands and the head; voice is also commonly affected. Tremor occurs mainly in association with action and is worse with anxiety. Therapy is recommended only in significantly affected individuals; it includes use of propranolol and primidone. Wilson disease is a rare cause of tremor in children. Diagnosis and management are discussed in Chapter 573.
 MYOCLONUS
MYOCLONUS
Myoclonus consists of sudden, brief, shocklike movements. These movements may be “positive” or “negative” (asterexis). Positive myoclonus results in contracture of a muscle or multiple muscles. In asterexis, there is a brief loss of muscle tone and then contraction of the muscles resulting in a flapping-type motion. Myoclonus can be rhythmic or non-rhythmic, focal, multifocal, or generalized, and can be activated by movement or sensory stimulation. Myoclonus is classified as physiologic, essential, epileptic, and symptomatic myoclonus (eTable 566.4  ).15
).15
Physiologic myoclonus occurs in normal people when falling asleep, during sleep and during waking. Epileptic myoclonus is associated with epileptiform activity on EEG. Essential myoclonus is a chronic condition of focal, segmental, or generalized jerking that is aggravated by action. Onset is between the first and second decade. A family history of myoclonus may be obtained, but sporadic cases occur. Treatment recommended is clonazepam, or several antiepileptic medications such as levetiracetam, clonazepam, valproic acid, primidone, piracetam, and acetazolamide.
 TICS AND TOURETTE SYNDROME
TICS AND TOURETTE SYNDROME
Tics are involuntary, sudden, rapid, repetitive, nonrhythmic, stereotyped movements (motor tics) or sounds (vocal tics). Tics can present in a variety of forms and have different degrees of severity and duration. Tics can present as simple tics such as eye blinking or shoulder shrug, or complex such as a stereotyped sequence of movements (touching, hitting, and smelling). Complex vocalization includes syllables, phrases, echolalia (repeating other people’s words), palilalia (repeating one’s own words), or coprolalia (obscene words).16 Tics have certain characteristic features such as a fluctuating course, suggestible nature, exacerbation during periods of anticipation and emotional upset, reduction when absorbed in activities, premonitory sensation (in a form of an urge, impulse, tension), and voluntary suppressibility.
Tics can be divided into 2 major categories based on their duration: transient (present less than 12 months) and chronic (present for more than 12 months).17 Based on etiology, tics can also be divided into primary (idiopathic), such as Tourette syndrome, chronic motor tic disorder, chronic vocal tic disorder, and transient tic disorder, and secondary, such as related to infection, drugs, toxins, and developmental and chromosomal disorders18 (eTable 566.5 ). Drug- or toxin-induced tics can be caused by stimulants, carbamaze-pine, steroids, neuroleptics, and carbon monoxide. Parainfectious tics can occur after streptococcus infection (PANDAS: postinfectious autoimmune neuropsychiatric disorder associated with streptococcus infection). Tics can also be seen with certain genetic and neurodegenerative disorders.
). Drug- or toxin-induced tics can be caused by stimulants, carbamaze-pine, steroids, neuroleptics, and carbon monoxide. Parainfectious tics can occur after streptococcus infection (PANDAS: postinfectious autoimmune neuropsychiatric disorder associated with streptococcus infection). Tics can also be seen with certain genetic and neurodegenerative disorders.
Tourette syndrome (TS) represents only 1 entity in a spectrum of tic disorder. TS presents with multiple motor and at least 1 vocal tic, a waxing and waning course, with tics evolving in a progressive manner; the presence of tic symptoms for at least 1 year, the onset of symptoms before age 21, the absence of precipitating illnesses or medication, and the observation of tics by a medical professional.19
Tics are very common in children, more often seen in boys, and most of them resolve spontaneously and do not require treatment unless they interfere with school and social interactions. The prevalence of moderately severe cases is estimated as 1 to 10/1000 children and adolescents.20,21 Onset of tics occurs any time from 2 to 15 years of age. Neck muscles are often affected first. New tics can occur in addition to or in place of an existing tic and can affect the head, eyes, and face. Common tics include eye blinking, grimacing, lip smacking, and shrugging of 1 or both shoulders. Verbal tics include a clearing of the throat, snorting or sniffing noise, and coughike noise.
Many patients with tics have associated psychopathology that includes attention deficit disorder (ADD) and obsessive-compulsive disorder (OCD).22 ADD is characterized by hyperactivity, short attention span, restlessness, poor concentration, and impaired impulse control. OCD is characterized by ritualistic behavior and thoughts. Typical OCD behavior includes placing objects repetitively in a certain place, washing hands, obsessive thoughts of violence, and counting objects.
The cause of tics is unknown; however, several family members may be affected, suggesting a genetic component.23 Convincing evidence exists that cortico-striatalthalamo-cortical pathways are involved in the expression of TS and tics and accompanying neuropsychiatric problems.24,25 The precise pathophysiologic location of tics remains unknown.
Diagnosis of tics and TS is based on the history and clinical examination. It is important for the examiner to observe the patient or a videotape of the tics. A complete neurological examination is essential to exclude other organic etiologies of abnormal movement. Brain MRI and EEG are normal.
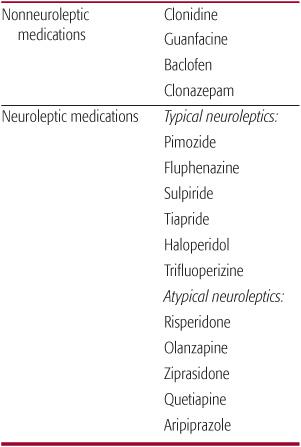
Many tics do not require therapy. The approach to treatment should be directed to improving quality of life. If tics are severe, the treatment should try to eliminate them during school hours; the dose of medication can be decreased or discontinued during summer months or when the child is not under stress. If pharmacological therapy is indicated, 2 categories of medications are available: nonneuroleptic drugs for milder tics and typical/atypical neuroleptics for more severe tics26 (Table 566-2). Botulinum toxin27,28 has been used in treating motor and vocal tics, and deep brain stimulation surgery (DBS) was also recently proposed for use in severe disabling cases of TS.29,30
Symptoms of OCD are treated with selective serotonin reuptake inhibitors (SSRIs) such as fluoxetine, sertraline, citalopram, or tricyclic antidepressants. Behavioral therapy can be used to reduce the frequency of compulsions.31 ADD is treated with CNS stimulants such as methylphenidate or dextroamphetamine,32 α2-adrenergic agonists (clonidine, quanfacine), and tricyclic antidepressants.33
 STEREOTYPIES
STEREOTYPIES
Stereotypies are broadly defined as involuntary, patterned, coordinated, repetitive, nonreflexive movements that are purposeless and occur in exactly the same manner with each repetition (eg, recurrent arm flapping, hand waving, finger wiggling, body rocking, leg shaking).34 Stereotypies can be classified as physiologic or pathologic. Physiologic stereo-typies (rocking, thumb sucking, lip or cheek biting) imply that the individual is otherwise normal and suggest a developmental problem that may improve with maturation of the nervous system. Stereotypies begin in infancy or early childhood; movements last seconds to minutes, tend to occur in clusters, appear many times per day, have a fixed pattern, and are associated with periods of excitement, stress, fatigue, or boredom. Movements can be simple or complex and can be suppressed by a sensory stimulus or distraction. The underlying pathophysiologic mechanism is unknown. Therapy is usually not indicated.
Pathologic stereotypies occur in patients with mental retardation, autism, Rett syndrome, neuroacanthocytosis, schizophrenia, obsessive-compulsive disorder, Tourette syndrome, and tardive dyskinesia.35
Table 566-2. Medication for Tics Nonneuroleptic medications
HYPOKINETIC MOVEMENT DISORDERS
 PARKINSONISM
PARKINSONISM
Parkinsonism is a syndrome manifested by a combination of 6 cardinal features: tremor at rest, rigidity, bradykinesia, loss of postural reflexes, flexed posture, and freezing.36 Juvenile Parkinson disease refers to a condition in which symptoms occur before age 20 years. The most common form of parkinsonism presenting at a young age is autosomal-recessive juvenile parkinsonism (AR-JP) due to mutation in the parkin gene on chromosome 6q25.2-27 (PARK 2). These individuals classically present with dys-tonic gait, levodopa-responsive dystonia, symmetric onset, marked sleep benefit, and early levodopa-induced dyskinesia. Patients with this form of parkinsonism seem to have a slower disease course.37 Other causes of juvenile parkinsonism include Parkinson disease due to PARK1, PARK 3, PARK 6, PARK 7, dopa-responsive dystonia, SCA 2, SCA3, and rapid-onset-dystonia-parkinsonism (DYT12), which can have its onset as early as the second decade.38
REFERENCES
See references on DVD.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


