Meconium Syndromes and Cystic Fibrosis
Sonya R. Walker
Edward M. Barksdale Jr.
Department of Surgery, University of Pittsburgh School of Medicine, Division of Pediatric Surgery, Children’s Hospital of Pittsburgh, Pittsburgh, Pennsylvania 15213.
Department of Surgery, University of Pittsburgh School of Medicine, Division of Pediatric Surgery, Children’s Hospital of Pittsburgh, Pittsburgh, Pennsylvania 15213.
The meconium syndromes encompass multiple gastrointestinal disorders leading to intestinal obstruction from meconium or foreign material. This group of diseases, including meconium ileus (MI; simple and complex), meconium peritonitis, meconium plug syndrome, and MI equivalent, share various overlapping aspects in pathophysiology, clinical presentation, diagnostic techniques, and management. The most common of these gastrointestinal meconium disorders, and their critical pathogenetic and molecular relationship to cystic fibrosis (CF), are discussed in this chapter.
MECONIUM ILEUS
History
First described by Landsteiner in 1905, MI is the newborn bowel obstruction caused by inspissated meconium, and associated with cystic degeneration and fibrosis of the pancreas. Pancreatic exocrine deficiency leading to the abnormal production of thick, viscous intestinal mucus was ultimately shown to be the primary cause of atypical meconium development (1,2,3). In 1936, Fanconi first reported CF, noting the association of pancreatic insufficiency and chronic pulmonary disease (4). Shortly thereafter in 1938, Anderson established the critical link between CF and MI, characterizing the histologic similarities between the bowel and pancreatic tissues (5). Evolution in surgical and medical therapies closely paralleled understanding of the pathophysiology of the disease. Prior to the mid-twentieth century, MI was generally fatal in newborns. In 1948, Hiatt and Wilson reported the first successful surgical treatment of MI using enterotomy and saline irrigation (6). Although several other surgical procedures were subsequently developed and significantly improved the survival for neonates with complicated MI, the concepts developed by Hiatt and Wilson are still today the basis of therapy for uncomplicated MI. In 1969, Noblett introduced a technique of nonoperative management of neonates with simple MI using hyperosmolar diatrizoate enema (7). Various modifications have been developed; however, the dilute water-soluble contrast enema remains the nonoperative standard of care for uncomplicated MI.
Epidemiology
Occurring primarily in white populations, the meconium syndromes are closely linked to CF. CF is one of the most common serious genetic diseases in whites. Approximately 5% to 6% of white individuals are carriers of the genetic defect (3,8). The incidence is about 1 in every 2,500 childbirths in the United States (9,10,11). CF is transmitted as an autosomal recessive trait, thus both parents must be heterozygotes for the gene in order to have an affected child. Each offspring has a one in four chance of developing the disease. MI will be the initial clinical manifestation of this disorder in 10% to 20% of affected infants. Ten to 30% of patients diagnosed with MI have a family history of CF. In families in which the first CF child has had MI, 29% of subsequent siblings with CF have MI, compared with 6% in families in which the first child with CF did not have MI (12,13). The number of males and females affected is almost equal. MI is uncommon in premature infants.
Genetics
Localization of the CF gene along with identification of the most prevalent genetic mutation was initially described in 1989 (14,15). Located on the long arm of chromosome 7, the gene has more than 800 known mutations at present (16,17). The most common mutation, ΔF508, a three base pair deletion from the gene, is responsible for approximately 70% of clinical cases (18,19). The gene encodes the cystic fibrosis transmembrane regulator (CFTR), which
is a cyclic adenosine monophosphate (cAMP)-induced chloride channel protein that resides in the apical membrane of epithelial cells (20). CFTR serves as a chloride channel activated by cAMP that regulates transepithelial fluid balance. Specifically, CFTR abrogates an amiloride-sensitive epithelial chloride channel, while inducing an alternate chloride channel (21) (Fig. 77-1). Mutations in this 1480 amino acid protein result in defective chloride transport in the apical membrane of epithelial cells of the respiratory, gastrointestinal, biliary, pancreatic, and reproductive systems. Types of defects fall into four or five major pathophysiologic categories that are based on how they disrupt CFTR synthesis and function (Table 77-1). CFTR synthesis occurs in the cell nucleus and undergoes folding and packaging in the endoplasmic reticulum prior to transport to the epithelial cell membrane. The membrane-bound CFTR regulates conductance through the chloride channels by responding to the appropriate signals (Fig. 77-2). The most common mutation, ΔF508, a class 2 defect, results in CFTR protein degradation in the endoplasmic reticulum prior to folding and trafficking. Patients who are homozygotes for this mutation typically develop significant pancreatic insufficiency, but may have variable pulmonary disease. The genetic basis of MI is unknown.
is a cyclic adenosine monophosphate (cAMP)-induced chloride channel protein that resides in the apical membrane of epithelial cells (20). CFTR serves as a chloride channel activated by cAMP that regulates transepithelial fluid balance. Specifically, CFTR abrogates an amiloride-sensitive epithelial chloride channel, while inducing an alternate chloride channel (21) (Fig. 77-1). Mutations in this 1480 amino acid protein result in defective chloride transport in the apical membrane of epithelial cells of the respiratory, gastrointestinal, biliary, pancreatic, and reproductive systems. Types of defects fall into four or five major pathophysiologic categories that are based on how they disrupt CFTR synthesis and function (Table 77-1). CFTR synthesis occurs in the cell nucleus and undergoes folding and packaging in the endoplasmic reticulum prior to transport to the epithelial cell membrane. The membrane-bound CFTR regulates conductance through the chloride channels by responding to the appropriate signals (Fig. 77-2). The most common mutation, ΔF508, a class 2 defect, results in CFTR protein degradation in the endoplasmic reticulum prior to folding and trafficking. Patients who are homozygotes for this mutation typically develop significant pancreatic insufficiency, but may have variable pulmonary disease. The genetic basis of MI is unknown.
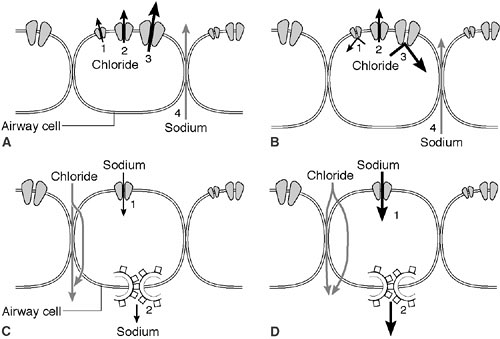 FIGURE 77-1. Chloride and sodium transport in normal and cystic fibrosis (CF) epithelial cells. (A) Epithelial cell (normal) with three types of chloride channels: 1, calcium dependent; 2, outwardly rectifying channel (ORCC); and 3, CF transmembrane regulator (CFTR), most important. Sodium passes between epithelial cells into the lumen. (B) CF epithelial cell with dysfunctional CFTR preventing chloride excretion. Calcium-dependent channel does not open, and the ORCC works minimally. (C) Epithelial cell (normal) sodium absorption. (D) CF epithelial cell with markedly upregulated sodium absorption. |
TABLE 77-1 CFTR Mutations and Their Role in Cystic Fibrosis Transmembrane Regulator Production and Function.a | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||
Pathogenesis
Cystic fibrosis is a systemic illness with diverse clinical presentations in various exocrine glands throughout the body. Abnormally thick and viscous mucous secretions may obstruct the bronchoalveolar tree, pancreatic ducts, and intestinal tract. Nasal mucus membranes, sweat and salivary glands, liver, and reproductive organs are also frequently affected. Common manifestations of this disease include pancreatic insufficiency in 90%, diabetes mellitus in 20%, obstructive biliary disease in 15–20%, and meconium ileus in 10–20% of all patients. Azoospermia occurs in nearly all affected males (22). Exocrine glands throughout the body are affected by this chloride transport defect. Abnormally thick and viscous mucus secretions obstructing the bowel, the pancreatic ducts, and air passages in the lung cause the most severe clinical manifestations.
The secretion of hyperviscous mucus and subsequent intestinal obstruction begins prenatally. The meconium
involved in MI is typically low in water content, minerals, and protein-bound carbohydrate and has several biochemical abnormalities, including elevated albumin, mucoprotein, and calcium, as well as low trypsin levels (23,24). The decreased carbohydrate and increased protein concentrations likely account for the hyperviscosity of the meconium (25,26). Neonates with MI and CF have a meconium protein content of 80% to 90%, compared with 7% in normal infants (27). Most of this extra protein has been shown to be albumin (28). The high viscosity and tenacity of the meconium result from a combination of a hyperviscous mucus secreted by abnormal intestinal glands, abnormal concentrating mechanisms of the proximal small intestine, and pancreatic exocrine deficiency in utero. Infants with MI and CF have severely affected intestinal glands, but mild pancreatic disease. It is believed that the intestinal glandular abnormality may be the primary precipitating factor for the development of meconium ileus, with the pancreatic disease playing a secondary role (29). The distal intestinal lumen becomes obstructed with meconium, leading to proximal bowel dilatation. Inspissated meconium located in the distal ileum and proximal colon develops into hard pellets due to the increased absorption of fluid from the stool. Disuse atrophy in utero results in a small-caliber colon or microcolon.
involved in MI is typically low in water content, minerals, and protein-bound carbohydrate and has several biochemical abnormalities, including elevated albumin, mucoprotein, and calcium, as well as low trypsin levels (23,24). The decreased carbohydrate and increased protein concentrations likely account for the hyperviscosity of the meconium (25,26). Neonates with MI and CF have a meconium protein content of 80% to 90%, compared with 7% in normal infants (27). Most of this extra protein has been shown to be albumin (28). The high viscosity and tenacity of the meconium result from a combination of a hyperviscous mucus secreted by abnormal intestinal glands, abnormal concentrating mechanisms of the proximal small intestine, and pancreatic exocrine deficiency in utero. Infants with MI and CF have severely affected intestinal glands, but mild pancreatic disease. It is believed that the intestinal glandular abnormality may be the primary precipitating factor for the development of meconium ileus, with the pancreatic disease playing a secondary role (29). The distal intestinal lumen becomes obstructed with meconium, leading to proximal bowel dilatation. Inspissated meconium located in the distal ileum and proximal colon develops into hard pellets due to the increased absorption of fluid from the stool. Disuse atrophy in utero results in a small-caliber colon or microcolon.
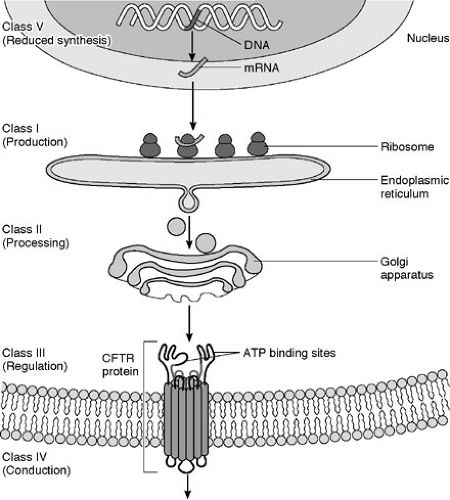 FIGURE 77-2. Cystic fibrosis gene and protein. CFTR, cystic fibrosis transmembrane regulator; ATP, adenosine triphosphate. |
Pancreatic injury begins prenatally and continues throughout the lifetime of the individual. The progressive obstruction of pancreatic ducts leads to acinar atrophy, decreased exocrine secretions, and eventually, fibrosis and fatty changes of the exocrine gland (30,31). By early infancy, approximately 85% to 90% of CF patients have advanced pancreatic lesions that result in an absence of pancreatic enzymes in the duodenum (32). Autopsy studies have revealed that CF patients without MI have a pattern of progressive pancreatic involvement with age. In contrast, patients with MI generally have mild pancreatic involvement and more severe intestinal glandular disease (29). The glandular changes include dilation of the crypts of Lieberkuhn, accumulation of intraluminal secretions, and flattening of the epithelial lining cells.
At birth, the lungs are normal in CF patients. However, progressive and diffuse pulmonary disease develops as a result of mucus plugging of the small airways and secondary infection. The sweat sodium and chloride levels are elevated from birth. These levels are unrelated to the severity or distribution of organ involvement. The sweat electrolyte abnormality is caused by the impermeability of the epithelia to chloride ions. The chloride is unable to follow as the sodium is actively pumped out of luminal fluids. Sodium tends to be retained in the lumen of the apocrine glands by this mechanism (33) (Fig. 77-1).
Classification
Simple Meconium Ileus
MI is usually classified as either simple (uncomplicated) or complex (complicated). In simple cases, the distal ileum
becomes obstructed by abnormal meconium as a result of a simple obturation. The proximal ileum is thickened, dilated, and packed with tarlike meconium. Distal to the site of obstruction, the bowel is collapsed and contains concrete, puttylike pellets of gray, inspissated meconium. The meconium in the distal bowel sometimes has a beadlike appearance. Due to the proximal obstructive disease, there is a microcolon, which is poorly developed and may contain small pellets of meconium. There is no classic histologic appearance for MI; however, tissue sections of the bowel may show intracellular inclusion bodies and intestinal crypts filled with inspissated meconium (Fig. 77-3).
becomes obstructed by abnormal meconium as a result of a simple obturation. The proximal ileum is thickened, dilated, and packed with tarlike meconium. Distal to the site of obstruction, the bowel is collapsed and contains concrete, puttylike pellets of gray, inspissated meconium. The meconium in the distal bowel sometimes has a beadlike appearance. Due to the proximal obstructive disease, there is a microcolon, which is poorly developed and may contain small pellets of meconium. There is no classic histologic appearance for MI; however, tissue sections of the bowel may show intracellular inclusion bodies and intestinal crypts filled with inspissated meconium (Fig. 77-3).
Complex Meconium Ileus
In utero, simple obturation obstruction of the bowel may progress to complicated MI. Intestinal necrosis or atresia, meconium peritonitis, pseudocyst, or all these conditions may occur in cases of complex MI. Most often, this is related to volvulus or ischemia of the dilated segment of the proximal ileum. Depending on the time at which the volvulus occurs and the in utero evolution of the process, the complicated form of this disease may result in intestinal atresia, perforation, or meconium peritonitis. Bacterial peritonitis results from postnatal volvulus and perforation, but occurs infrequently (Fig. 77-4).
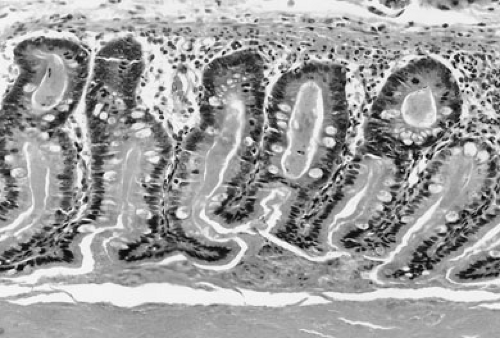 FIGURE 77-3. Inspissated mucus in meconium ileus. Histologic section of bowel showing dilated crypts filled with eosinophilic, inspissated mucus. |
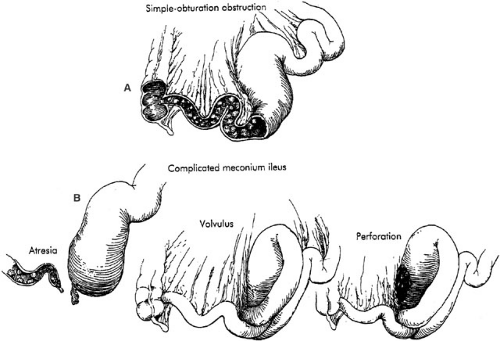 FIGURE 77-4. Complex meconium ileus (MI). (A) Simple MI with obstruction of the ileum by inspissated meconium pellets. The bowel proximal to the obstruction is dilated, in contrast to the small caliber of the colon distal to the obstruction. (B) Depiction of three variations of complex MI (atresia, volvulus, and perforation). (From Rescorla FJ, Grosfeld JL. Contemporary management of meconium ileus. World J Surg 1993;17:318, with permission.) |
Meconium Peritonitis
Meconium peritonitis was first reported by Morgagni in 1761 in De Sedibus et Causis Morborum as an aseptic, chemical, or foreign-body reaction resulting from prenatal spillage of meconium into the peritoneal cavity due to intestinal perforation (34,35). Ischemic necrosis and perforation result from the large meconium bolus located
proximal to the obstruction. A segmental volvulus often occurs when the meconium-filled segment of redundant ileum twists. The narrow base of the volvulus may lead to ischemic necrosis of the bowel and result in isolated or multiple ileal atresias, formation of a stricture, or perforation. The perforation is typically secondary to segmental volvulus followed by gangrene with focal perforation (Fig. 77-4). Infants with complicated MI may present with intestinal perforation in the form of either free or encysted peritonitis. Three pathologic types of meconium peritonitis have been described, including generalized, fibroadhesive, and cystic (36). Generalized meconium peritonitis with ascites develops if the perforation and meconium extravasation occur in the late perinatal or early neonatal period. Diffuse distribution of meconium throughout the peritoneal cavity initiates the inflammatory process. However, due to the brief duration, no calcification occurs. The adhesions between bowel loops are more fibrinous, rather than fibrous, in generalized peritonitis. Most commonly, however, fibroadhesive meconium peritonitis occurs. When the perforation has occurred early enough prior to delivery, dense adhesions and calcification occur. Intraperitoneal meconium may form calcifications within 48 hours of perforation (37). The meconium contains digestive enzymes that induce an intense chemical peritonitis, initiated by the sterile meconium. Subsequently, dense, fibrous adhesions and agglutination of the bowel may result. The calcified and fibrous adhesions are often effective in sealing the site of perforation and frequently obscure identification of the site. When the fibroblastic reaction has not been effective in sealing the site of perforation, meconium may continue to leak into the peritoneal cavity, resulting in cystic meconium peritonitis. Segmental volvulus with ischemic necrosis of the bowel along with extravasation and liquefaction of meconium may lead to development of a pseudocyst. The pseudocyst consists of densely adherent, partially necrotic loops of intestine surrounding liquefied meconium. Frequently, a calcified fibrous peel or wall envelops this mass. This cyst may fill with air after birth and may then be called giant cystic meconium peritonitis (38). Colonic perforation distal to the obstruction may result from excessive intraluminal pressure from a contrast enema; however, this is not generally a difficult event to recognize and differentiate from the previous lesions.
proximal to the obstruction. A segmental volvulus often occurs when the meconium-filled segment of redundant ileum twists. The narrow base of the volvulus may lead to ischemic necrosis of the bowel and result in isolated or multiple ileal atresias, formation of a stricture, or perforation. The perforation is typically secondary to segmental volvulus followed by gangrene with focal perforation (Fig. 77-4). Infants with complicated MI may present with intestinal perforation in the form of either free or encysted peritonitis. Three pathologic types of meconium peritonitis have been described, including generalized, fibroadhesive, and cystic (36). Generalized meconium peritonitis with ascites develops if the perforation and meconium extravasation occur in the late perinatal or early neonatal period. Diffuse distribution of meconium throughout the peritoneal cavity initiates the inflammatory process. However, due to the brief duration, no calcification occurs. The adhesions between bowel loops are more fibrinous, rather than fibrous, in generalized peritonitis. Most commonly, however, fibroadhesive meconium peritonitis occurs. When the perforation has occurred early enough prior to delivery, dense adhesions and calcification occur. Intraperitoneal meconium may form calcifications within 48 hours of perforation (37). The meconium contains digestive enzymes that induce an intense chemical peritonitis, initiated by the sterile meconium. Subsequently, dense, fibrous adhesions and agglutination of the bowel may result. The calcified and fibrous adhesions are often effective in sealing the site of perforation and frequently obscure identification of the site. When the fibroblastic reaction has not been effective in sealing the site of perforation, meconium may continue to leak into the peritoneal cavity, resulting in cystic meconium peritonitis. Segmental volvulus with ischemic necrosis of the bowel along with extravasation and liquefaction of meconium may lead to development of a pseudocyst. The pseudocyst consists of densely adherent, partially necrotic loops of intestine surrounding liquefied meconium. Frequently, a calcified fibrous peel or wall envelops this mass. This cyst may fill with air after birth and may then be called giant cystic meconium peritonitis (38). Colonic perforation distal to the obstruction may result from excessive intraluminal pressure from a contrast enema; however, this is not generally a difficult event to recognize and differentiate from the previous lesions.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


