Lung
Heidi J. Pinkerton
Keith T. Oldham
Division of Pediatric Surgery, Children’s Hospital of Wisconsin, Milwaukee, Wisconsin 53226.
Division of Pediatric Surgery, Medical College of Wisconsin, Children’s Hospital of Wisconsin, Milwaukee, Wisconsin 53226.
Infants and children manifest an extraordinary diversity of congenital and acquired abnormalities of the lung. Although none is truly common, all the surgical conditions presented in this chapter are important because physiologic lung dysfunction is a potential consequence. Because impaired respiratory gas exchange can be life threatening, it is of fundamental importance that those who care for infants and children are familiar with these lesions. A full understanding of respiratory physiology requires consideration of the chest wall, diaphragm, and airways, in addition to the lungs. This book considers each component separately for reasons of organizational and educational expediency. The reader is referred to other chapters for these specific related discussions, and also to Chapter 11, which considers respiratory physiology in detail.
EMBRYOLOGY AND ANATOMY
Lung Development
By the end of the third week of gestation, the laryngotracheal groove is discernible in the ventral aspect of the proximal foregut in human embryos. This groove develops into a tracheal diverticulum, a primordium that elongates caudally and lies ventral and parallel to the dorsal foregut, the primitive esophagus. By the middle of week 6, the trachea has undergone symmetric distal division into the left and right mainstem bronchi. Mesenchymal proliferation follows in the adjacent mediastinum and is necessary for normal lung organogenesis. This mesoderm is ultimately the source of cartilage, smooth muscle, and connective tissue to the developing lungs. Progressive bronchial branching follows, and by the seventh gestational week, three to five orders of bronchi are present. Tracheal and esophageal separation are normally complete at this time (1). All major bronchial beds are present by 8 to 9 weeks’ gestation, when closure of the pleural peritoneal canal completes formation of the diaphragm. By this time, segmental and lobar lung development is complete. After about 16 weeks’ gestation, the primary events in fetal lung development are related to the successive formation of terminal airways and alveoli—the critical sites for respiratory gas exchange. This process is of profound importance in understanding the physiologic consequences of mass lesions in the fetal thorax (2) (see Chapters 11 and 58). With regard to understanding the specific physiologic response to pulmonary surgery in infants, several points bear reiteration. Alveoli undergo highly significant maturation and development during the third trimester and during early postnatal life. Both the number and size of alveoli increase during this time. Most of this process is completed by 2 to 4 years of age, although it continues until about age 8 (2,3,4). Because of this age-related potential for compensatory lung growth, the tolerance of pulmonary resection in infants and children is generally good.
Normal Anatomy
Key anatomic relations of the pulmonary hilar to adjacent structures are summarized in Fig. 61-1. The right lung is normally larger than the left, with an upper, middle, and lower lobe each supplied by its respective bronchus. The left lung is formed by upper and lower lobes similarly situated, and the lingula is analogous to the right middle lobe. The tracheal bifurcation is at the fourth or fifth vertebral body by the time of birth. The right mainstem bronchus is normally larger, more vertical, and about one-half the length of the left. These anatomic features account for the preferential drainage of endobronchial material or foreign bodies into the right lung, particularly the right lower lobe.
The circulation on which respiratory gas exchange depends is derived from the pulmonary artery and its branches. The circulation that provides nutritive support
for the lung parenchyma and the bronchi, however, is systemic. The main left bronchus and its dependent airways are supplied by two bronchial arteries that arise from the anterior surface of the descending thoracic aorta. On the right, a single bronchial artery is usually present and is derived from either the third right intercostal artery or the more superior of the two left bronchial arteries. Intramural collateral flow from the trachea to the bronchi is derived from the inferior thyroid arteries, which are supplied by the thyrocervical trunks of the subclavian arteries. Venous bronchial drainage is into the azygous and hemiazygous systems. Generally, both arteries and veins follow the segmental and lobar architecture of the bronchi.
for the lung parenchyma and the bronchi, however, is systemic. The main left bronchus and its dependent airways are supplied by two bronchial arteries that arise from the anterior surface of the descending thoracic aorta. On the right, a single bronchial artery is usually present and is derived from either the third right intercostal artery or the more superior of the two left bronchial arteries. Intramural collateral flow from the trachea to the bronchi is derived from the inferior thyroid arteries, which are supplied by the thyrocervical trunks of the subclavian arteries. Venous bronchial drainage is into the azygous and hemiazygous systems. Generally, both arteries and veins follow the segmental and lobar architecture of the bronchi.
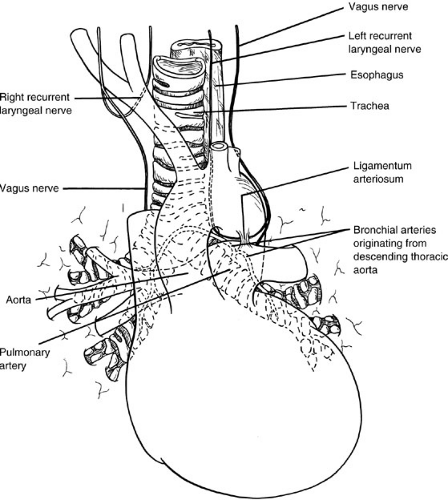 FIGURE 61-1. Key anatomic relations of the structures of the pulmonary hilum. |
In large airways at the microscopic level, respiratory epithelium is ciliated, pseudostratified columnar epithelium. Smooth muscle demarcates the basal extent of the mucosa. Mucus-producing glands are apparent in the submucosa. Parasympathetic ganglion cells are abundant, and cartilage is present. Cartilage is an essential structural element for maintaining expiratory patency of the major conducting airways. Proceeding distally in the tracheobronchial tree, luminal size diminishes, and certain features are lost. In particular, as the bronchus becomes a bronchiole, cartilage is lost, the glands disappear, and the epithelium becomes simple, columnar, and ciliated. Smooth muscle and elastic tissues remain. More distally, these elements drop out of the terminal bronchiole as the need to conduct large volumes of air diminishes. Eventually, at the level of the respiratory bronchiole and the alveoli, where gas exchange occurs, capillaries and epithelial cells are juxtaposed intimately. Most oxygen and carbon dioxide diffusion actually occurs across a membrane composed only of thin cytoplasmic extensions of the type I epithelial cell and the capillary endothelial cell, bound by a common basement membrane. This is only nanometers in thickness.
Parenchymal lung lesions in infants and children are divided in the following discussion into those that are congenital and those that are acquired. Anomalies that result from disordered organogenesis during foregut development are reviewed in detail as follows (5,6,7). Certain conditions, such as cystic fibrosis (CF) and bullous disease, may have both congenital and acquired features, and these individual aspects are also reviewed in the following sections.
CONGENITAL DISORDERS
Congenital Lobar Emphysema
Congenital lobar emphysema, or congenital lobar overinflation, refers to the abnormal postnatal collection of air within a lobe of the lung that is otherwise anatomically normal. This condition is characterized by expiratory air-trapping within the affected lobe, resulting in lobar parenchymal distention. Compression of adjacent normal lung and mediastinal structures is expected, and physiologic impairment of gas exchange is common. The process is classically the result of developmental deficiency of the cartilage that supports the bronchus to the involved lobe, resulting in focal bronchial collapse and obstruction to expiratory air flow. This specific defect, however, is demonstrable in only one-third to two-thirds of surgically resected emphysematous lobes (8). The remainder of these infants and children have a variety of partially obstructing bronchial lesions. Some are endobronchial in origin and potentially reversible (e.g., viscid secretions, mucous plugs, or granulation tissue). Some are the result of extrinsic compression of the bronchus with partial obstruction. Causes include mediastinal lymphadenopathy; adjacent vascular structures, such as an aberrant or enlarged pulmonary artery or ductus arteriosus; mediastinal cysts or tumors that are bronchogenic in origin; or other congenital or acquired mediastinal lesions with an intimate hilar relation. For these reasons, preresection bronchoscopic evaluation is recommended with the expectation that reversible bronchial obstruction be corrected before sacrificing a lung lobe that is otherwise normal.
In addition, developmental abnormalities of the alveoli or the terminal airways may be associated with the clinical findings of congenital lobar emphysema. Of note in this regard is the finding of polyalveolar morphology, a descriptive histologic term that refers to a substantial and abnormal increase in the number of alveoli present. In this circumstance, postnatal air-trapping occurs within these many alveoli.
Congenital lobar emphysema is a rare lesion, with a 2:1 or 3:1 male predominance. It is most common in the white population. Unilobar involvement is the rule, with affected sites distributed in the following manner: left upper lobe, 40% to 50%; right middle lobe, 30% to 40%; right upper lobe, 20%; lower lobes, 1%; and multiple sites the remainder (7,8) (Fig. 61-2). Congenital lobar emphysema is associated with congenital heart disease or abnormalities of the great vessels in about 15% of infants (9,10,11). Indeed, extrinsic bronchial compression from vascular structures appears to be a common etiologic problem in this circumstance. For this reason, screening echocardiography is appropriate in all infants with congenital lobar emphysema.
Affected infants usually do not have symptoms at birth. With the onset of extrauterine life and spontaneous respiration, air-trapping and progressive lobar distention develop. Initial clinical symptoms are generally tachypnea and dyspnea, followed by cyanosis if oxygenation is sufficiently impaired. A cough or wheezing may also be present, but this is of little specificity. About one-half of affected infants develop symptoms in the first few days of life; the remainder develop symptoms within the first 6 months. Older infants and children may have few or no symptoms. Infants may have rapidly progressive respiratory failure, with up to 10% to 15% of patients requiring emergency thoracotomy. Generally, the clinical progression is slower, and some patients remain without symptoms.
The clinical presentation may be one of progressive respiratory distress; therefore, an affected infant may become increasingly agitated, anxious, and tachypneic. These normal responses to hypoxemia exacerbate the air-trapping phenomenon as the peak inspiratory and expiratory pressures escalate. In particular, focal bronchial collapse occurs with excessive expiratory effort; as this develops, the lobar emphysema worsens, and further compromise in gas exchange results. Likewise, positive-pressure ventilation can induce acute lobar distention with potentially catastrophic respiratory decompensation or mediastinal displacement. The physiologic derangements may be indistinguishable from those of tension pneumothorax. This is an important consideration during endoscopic evaluation of the endobronchial tree. Particularly in infants with preoperative symptoms, the surgeon must be prepared to decompress the thorax by emergent thoracotomy and then to proceed with definitive lobectomy.
Congenital lobar emphysema is typically found in term infants, but acquired emphysematous disease in preterm infants is common. The cause of this latter problem is multifactorial, including barotrauma from positive-pressure ventilation, oxygen toxicity, and lung immaturity. It is often seen in conjunction with, and as a complication of, bronchopulmonary dysplasia. Unlike congenital lobar emphysema, multiple areas of focal hyperinflation and interstitial emphysema are often present. Unilobar right lower lobe involvement is also common, probably as a consequence of endotracheal tube positioning, which selectively ventilates the right mainstem bronchus. These characteristics help differentiate congenital and acquired disease.
Physical findings of congenital lobar emphysema may include an asymmetric thorax, a shift in the apical cardiac impulse to the contralateral side, and focal hyperresonance and diminished breath sounds over the affected lobe. None of these findings, however, has the necessary sensitivity and specificity to demonstrate the precise nature of the problem.
The diagnosis is best established by plain chest radiograph (Fig. 61-2). Typical findings include lobar hyperinflation, contralateral shift of the mediastinum and trachea, compression or even lobar atelectasis of adjacent lung, and flattening of the ipsilateral hemidiaphragm. If these
findings are all present, there is no need for additional imaging studies. Differentiating this presentation from tension pneumothorax is essential. The latter is characterized by collapse of the entire affected lung into the hilum. In contrast, although lobar emphysema can be dramatic in its radiographic appearance, adjacent compressed lung can almost always be discerned, most often the lower lobe at the base of the thorax. In addition, the occasional congenital cystic adenomatoid malformation (CCAM) with a single large cystic component can be mistaken for lobar emphysema. Because lobar emphysema rarely involves the lower lobes, this is an important differentiating feature. Nonetheless, the surgical management of these latter two lesions is similar so preoperative differentiation is less important than for tension pneumothorax, for which the treatment is different. As with most mass lesions in the chest, computed tomography (CT) and magnetic resonance (MR) imaging provide excellent anatomic information for infants with congenital lobar emphysema. These procedures are most helpful in elective situations when the diagnosis is in doubt. In addition, ventilation–perfusion scans have been employed to evaluate infants with lobar emphysema, particularly when the areas of involvement are multiple or the disease acquired (12). In this setting, specific areas of nonfunctional lung can be identified and resected if they appear to compromise adjacent normal lung.
findings are all present, there is no need for additional imaging studies. Differentiating this presentation from tension pneumothorax is essential. The latter is characterized by collapse of the entire affected lung into the hilum. In contrast, although lobar emphysema can be dramatic in its radiographic appearance, adjacent compressed lung can almost always be discerned, most often the lower lobe at the base of the thorax. In addition, the occasional congenital cystic adenomatoid malformation (CCAM) with a single large cystic component can be mistaken for lobar emphysema. Because lobar emphysema rarely involves the lower lobes, this is an important differentiating feature. Nonetheless, the surgical management of these latter two lesions is similar so preoperative differentiation is less important than for tension pneumothorax, for which the treatment is different. As with most mass lesions in the chest, computed tomography (CT) and magnetic resonance (MR) imaging provide excellent anatomic information for infants with congenital lobar emphysema. These procedures are most helpful in elective situations when the diagnosis is in doubt. In addition, ventilation–perfusion scans have been employed to evaluate infants with lobar emphysema, particularly when the areas of involvement are multiple or the disease acquired (12). In this setting, specific areas of nonfunctional lung can be identified and resected if they appear to compromise adjacent normal lung.
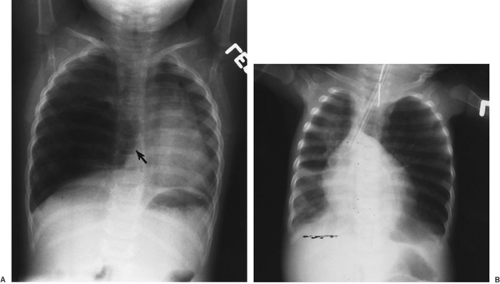 FIGURE 61-2. Congenital lobar emphysema involving the right middle lobe. (A) Herniation of the affected lobe across the midline has occurred (arrow), with compression of the adjacent right upper and right lower lobes. Mediastinal shift into the contralateral thorax is also apparent. (B) Similar abnormalities in a 4-month-old girl with involvement of the left upper lobe, the most common site of congenital lobar emphysema. |
Because the natural history of congenital lobar emphysema is often progressive and includes potentially life-threatening respiratory insufficiency, prompt surgical lobectomy is the treatment of choice for infants and young children. Because the underlying lesion is structural, medical treatment can be considered only a supportive adjunct in patients with symptoms. In patients without symptoms, particularly older children, this approach may be tempered reasonably because the likelihood of sudden decompensation in this circumstance is low. The rationale for routine endoscopic evaluation of the affected bronchus has been noted. The purpose is to identify and eliminate reversible endobronchial obstructions from secretions, mucous plugging, or granulation tissue. Clearly reversible endobronchial problems should be corrected without parenchymal lung resection.
Extrinsic bronchial compression is associated generally with a focal cartilaginous defect of the affected bronchus that is not adequately relieved by simple decompression.
Although congenital lobar emphysema results from a specific anatomic defect, reconstructive procedures, such as bronchoplasty or segmental bronchial resection and anastomosis, are generally inappropriate. The diminutive size of the infant bronchus and the possibility of nonfocal cartilaginous tracheobronchial defects present important technical obstacles to successful local reconstructive procedures. In addition, there is little reason to select this approach because the clinical results of lobectomy are generally excellent for this lesion (9,10,11,13).
Although congenital lobar emphysema results from a specific anatomic defect, reconstructive procedures, such as bronchoplasty or segmental bronchial resection and anastomosis, are generally inappropriate. The diminutive size of the infant bronchus and the possibility of nonfocal cartilaginous tracheobronchial defects present important technical obstacles to successful local reconstructive procedures. In addition, there is little reason to select this approach because the clinical results of lobectomy are generally excellent for this lesion (9,10,11,13).
Acquired emphysema is often seen in preterm infants with a multitude of other problems. Treatment is generally medical and supportive, with the natural history being one of slow resolution over a number of months. In the acute phase, selective ventilation of nonemphysematous areas of lung or the use of alternative strategies such as high-frequency oscillatory or jet ventilation can minimize the peak airway pressure, which is directly correlated to the formation of emphysema. These approaches can also help infants with congenital lobar emphysema if prolonged transport is necessary or there is delay in reaching the operating suite. Lobectomy may be beneficial in occasional selected patients with severe regional emphysematous disease; however, late death may result from associated bronchopulmonary dysplasia in these patients.
As noted, infants and children have an excellent response to lobectomy for congenital lobar emphysema (9,10,11,13). Even in those who are critically ill and require emergency thoracotomy, the physiologic response is a predictably prompt and dramatic return to normal after resection of the affected lobe. Mortality for this specific lesion is rare in a modern pediatric surgical environment. The general risks of thoracotomy and lung resection include morbidity related to anesthesia, empyema, pneumothorax, infection, bleeding, and bronchopleural fistula. These are not different than for any other neonatal thoracotomy and lobectomy, and are presented in detail in the section that deals with outcomes after lung resection. The cumulative incidence of these types of complications is about 5% to 10% in most modern pediatric surgical practices, although it has been as high as 20% to 40% in recent decades (7,9,10,11). Long-term pulmonary function is also predictably excellent after lobar resection, and this is discussed separately later. For infants with coexisting congenital heart disease, acquired pulmonary emphysema, or additional medical problems, the outcome is generally dictated by these other conditions.
In follow-up studies by Frenckner and Freyschuss, (14) actual lung volumes—residual volume, vital capacity, total lung capacity, and forced expiratory volume in 1 second (FEV1)—in patients who had undergone neonatal lobectomy for congenital lobar emphysema were 90% of predicted values, and no long-term functional impairment was reported. Infants who had undergone neonatal lobectomy for congenital lobar emphysema were evaluated as adults by McBride and colleagues in 1980 (13). Ipsilateral and contralateral lung volumes were found to be equal, despite the previous lobectomy. This appeared to be the result of compensatory tissue growth, not simply distention of residual lung parenchyma. In this latter study, perfusion was found to be equally distributed between the operated and nonoperated lungs. These patients demonstrated diminished expiratory flow rates compared with expected values (FEV1, 72% of predicted; maximal midexpiratory flow, 45% of predicted). These findings appear to result from disproportional growth between the conducting and the terminal airways during infancy. This concept does not diminish the excellent clinical prognosis for these infants, and is presented in detail at the end of this chapter.
Congenital Cystic Adenomatoid Malformation
Congenital cystic adenomatoid malformation (CCAM) is a term applied to a spectrum of lobar hamartomatous abnormalities of the lung. The pathologic definition requires an increase in terminal respiratory structures, usually bronchioles, in a glandular or adenomatoid pattern that is normally seen during organogenesis. It is suggested that developmental control of the lobar lung bud and the surrounding mediastinal mesenchyme is lost between 16 and 20 weeks’ gestation, giving rise to a lesion that is composed of multiple interconnected cysts that are disorganized and variably sized. Involvement is generally unilobar, and communication with the tracheobronchial tree is usually present. Three types of CCAM lesions were described by Stocker and colleagues (15) in 1977 and are illustrated in Fig. 61-3. Subsequently, others used the terms cystic, intermediate, and solid to categorize the different lesions observed with this malformation. More recently, Adzick and colleagues (16) defined these lesions as either macrocystic (greater than 5-mm cyst diameter) or microcystic (solid or less than 5-mm cyst diameter), by use of prenatal ultrasound examination to differentiate between the two. Because the natural history is dependent on morphologic type, the distinctions have more than semantic import. Clinical treatment and outcomes are presented later.
Both before and after parturition, the important physiologic consequences of CCAM result from mediastinal or normal lung compression by the mass lesion. Lesions of great size, particularly those that are microcystic or solid in composition, are potentially associated with in utero mediastinal displacement, hydrops fetalis, and fetal death. As many as one-third of all newborns with CCAM have evidence of fetal hydrops at delivery. It is now routine to establish the diagnosis of CCAM by prenatal ultrasound. The data for the fetus diagnosed with CCAM are more limited and controversial, but as many as 40% of fetuses with CCAM lesions will progress to hydrops and fetal demise, whereas 15% will spontaneously regress (17). Adzick and
colleagues (18) suggested that the appearance of anasarca or hydrops in fetuses with microcystic CCAM is an indication of impending fetal demise; they have reported neonatal survival after fetal lobectomy in a small number of highly selected patients (19). Macrocystic lesions appear less threatening in utero, and although some have been managed with prenatal thoracoamniotic shunting or aspiration, most of these infants can be successfully treated after delivery at term. In addition, it appears that a substantial number of macrocystic lesions, perhaps one-third, diminish in size during fetal development (20,21). This is an important area of active investigation. Appropriate selection of patients for prenatal therapy depends on accurate information defining the natural history of CCAM.
colleagues (18) suggested that the appearance of anasarca or hydrops in fetuses with microcystic CCAM is an indication of impending fetal demise; they have reported neonatal survival after fetal lobectomy in a small number of highly selected patients (19). Macrocystic lesions appear less threatening in utero, and although some have been managed with prenatal thoracoamniotic shunting or aspiration, most of these infants can be successfully treated after delivery at term. In addition, it appears that a substantial number of macrocystic lesions, perhaps one-third, diminish in size during fetal development (20,21). This is an important area of active investigation. Appropriate selection of patients for prenatal therapy depends on accurate information defining the natural history of CCAM.
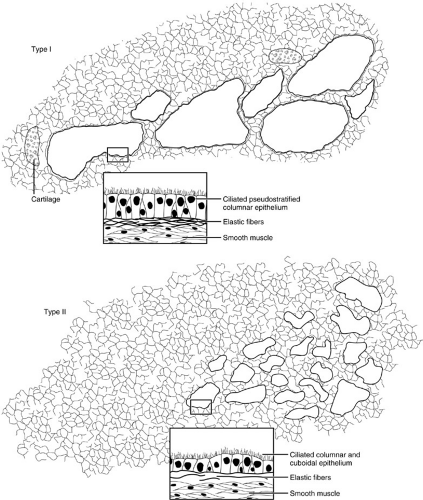 FIGURE 61-3. Congenital cystic adenomatoid malformation (CCAM) types. Type I lesions are composed of irregular cysts larger than 2 cm in diameter, elastic tissue is regularly present, and cartilage is present in 10% of lesions. The epithelium is ciliated, pseudostratified columnar epithelium. Relatively normal alveolar structures are adjacent to, or interspersed with, these cysts. Type II lesions have more and smaller cysts, less than 1 cm in diameter. The epithelium is ciliated cuboidal or columnar; there is less elastic tissue and no cartilage. The lesions blend into relatively normal lung parenchyma with large alveolus-like structures. Type III lesions occupy the entire affected lobe, and despite the CCAM nomenclature, there are no cystic spaces. Rather, masses of cuboidal epithelium line alveolus-like structures where no gas exchange can occur. (From Stocker JT, Madewell JE, Drake RM. Congenital cystic adenomatoid malformation of the lung. Hum Pathol 1977;8:155, with permission. |
Postpartum physiologic problems related to CCAM generally result from either pulmonary hypoplasia in newborns or inadequate tracheobronchial drainage with secondary infection in older infants and children. The former problem appears related to in utero compression and developmental arrest of the ipsilateral lung by the CCAM mass. In addition, some degree of contralateral pulmonary hypoplasia resulting from the shifted mediastinum is common. Pulmonary parenchymal hypoplasia and persistent pulmonary hypertension can result in acute respiratory failure in newborns. Severely affected infants with CCAM may have all the ventilatory instability of infants with congenital diaphragmatic hernias. This includes the potential need for conventional mechanical ventilation, high-frequency or jet ventilation, or extracorporeal life support. These issues are discussed in detail in Chapters 11 and 58. Although the spectrum of physiologic derangement includes both acute life-threatening respiratory failure and progressive newborn respiratory insufficiency, only about 30% of live-born infants with CCAM present in these ways. Many present with infectious pulmonary problems related to the persistent communication of the CCAM with the tracheobronchial tree. The abnormal lung parenchyma is exposed to environmental organisms, but lacks normal clearance mechanisms. This leads to a variety of infectious problems, such as recurrent pneumonia or lung abscess, or to more subtle chronic problems, such as failure to thrive. In contemporary practice, prenatal diagnosis yields a number of asymptomatic infants referred for treatment. In general, these infants should undergo elective lobectomy.
CCAM lesions are uncommon, representing about 30% to 40% of developmental lung bud anomalies in most reports. There is a slight male predominance and no apparent racial or geographic predilection. CCAM lesions are equally distributed between the left and right lobes, with bilateral disease being rare. Unilobar involvement is most common, with any lobe at risk, although there appears to be slight predilection for the lower lobes in most reports. Fortunately, multilobar disease when it occurs tends to be unilateral so surgical resection can be achieved by pneumonectomy, if necessary. A maternal history of polyhydramnios is common, and preterm delivery occurs in as many as one-half of these infants. Therefore, the many problems of preterm delivery may be superimposed.
Depending on the institutional environment, one-half or more of these lesions are detected and referred based on prenatal ultrasonography findings. As outlined earlier, about one-third of newborns with CCAM develop symptoms of tachypnea, dyspnea, cyanosis, or overt respiratory insufficiency in the first month of life. The remainder present with the consequences of pulmonary infection—one-half of these within the first year of life and the remainder at periods up to and including adulthood. Later presentations include recurrent or persistent pneumonia, lung abscess, pneumothorax, reactive airway disease, and failure to thrive, but not usually progressive respiratory insufficiency in older patients. Associated anomalies, including congenital heart disease, pectus excavatum, renal agenesis, skeletal anomalies, jejunal atresia, and others, have been reported, but the incidence is variable and may be no more than for the normal population (6,7).
Depending on the institutional environment, one-half or more of these lesions are detected and referred based on prenatal ultrasonography findings. As outlined earlier, about one-third of newborns with CCAM develop symptoms of tachypnea, dyspnea, cyanosis, or overt respiratory insufficiency in the first month of life. The remainder present with the consequences of pulmonary infection—one-half of these within the first year of life and the remainder at periods up to and including adulthood. Later presentations include recurrent or persistent pneumonia, lung abscess, pneumothorax, reactive airway disease, and failure to thrive, but not usually progressive respiratory insufficiency in older patients. Associated anomalies, including congenital heart disease, pectus excavatum, renal agenesis, skeletal anomalies, jejunal atresia, and others, have been reported, but the incidence is variable and may be no more than for the normal population (6,7).
The issues related to prenatal diagnosis have been discussed. The postnatal evaluation of infants with nonspecific respiratory symptoms is best begun with a plain chest radiograph. In infants with CCAM, however, the radiographic findings are variable. Images obtained shortly after birth may show retained fetal lung fluid within the lesion, and if it is a microcystic or solid lesion, this may not change with time. Macrocystic lesions tend to become aerated with ventilation, and the chest radiograph then has an area of air-filled cysts within the thorax. In infants, this appearance must be distinguished from congenital diaphragmatic hernia, particularly when the left side is involved. Although plain films alone are generally adequate, passage of a nasogastric tube into the stomach or an upper or lower gastrointestinal tract contrast study showing intrathoracic intestine may be helpful in distinguishing the two. Because the surgical approach is generally different for these two lesions, prospective distinction is important. Mediastinal displacement, compression of adjacent normal lung, and flattening of the intact ipsilateral diaphragm are also typical plain chest radiograph findings for CCAM. In older children with infectious complications, the findings are often less clear, and either CT with intravenous contrast or MR evaluation is necessary to provide definitive anatomic detail of the lesion (Fig. 61-4). Angiography has little or no role in the diagnosis of CCAM and other thoracic mass lesions in the modern environment because it has demonstrable risks and the information derived is available by less invasive means.
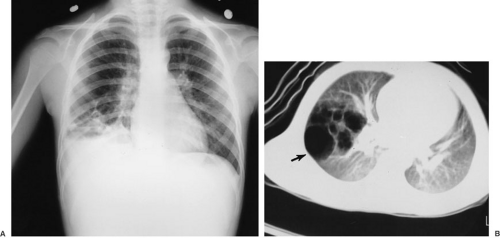 FIGURE 61-4. (A) Plain chest radiograph of a 9-year-old child who presented with fever, pleuritic chest pain, and cough. The lesion is an infected cystic adenomatoid malformation of the right lower lobe. (B) The lesion in A is shown on chest computed tomography scan after treatment with antibiotics and before surgical resection of the right lower lobe. (From Coran AG, Oldham KT. The pediatric thorax. In: Greenfield LJ, Mulholland MW, Oldham KT, et al. Surgery: scientific principles and practice. Philadelphia: JB Lippincott, 1993:813, with permission.) |
The principal goal of treatment for CCAM is to resect the area of abnormal lung promptly. Some carefully selected fetuses may benefit from prenatal intervention; however, concerns remain about the natural history of the lesions, appropriate patient selection, and the risk of preterm labor. Experience with this approach is limited and is
insufficient to be definitive. Generally, an in utero diagnosis and macrocystic disease are indications for sequential observation and delivery in a tertiary care environment where prompt thoracotomy and state-of-the-art critical care support are available. For the infant, treatment most often requires a thoracotomy with lobectomy. This can be life saving in critically ill newborns with mediastinal shift and normal lung compression from a ventilated and expanding CCAM. Fig. 61-5 demonstrates the relative size of a right lower lobe CCAM deliberately delivered from the thorax of an infant in extremis. The normal adjacent lung was allowed to ventilate, providing immediate physiologic relief before lobar resection. Because of the long-term risk of infectious complications, surgical resection is considered standard in older patients and in patients without symptoms. It is appropriate in the setting of an acute infectious process to treat a child preoperatively with systemic antibiotics to reduce acute inflammation. Long-term medical management, however, is not appropriate. Approximately 8% of primary lung malignant tumors and 4% of benign tumors are associated with cystic malformation of the lung, including CCAMs (22). To date, at least 24 reports of malignancy occurring within these and other congenital cystic lung lesions add further rationale for surgical resection (22,23,24,25). Pulmonary blastoma and rhabdomyosarcoma are the most common of these malignancies.
insufficient to be definitive. Generally, an in utero diagnosis and macrocystic disease are indications for sequential observation and delivery in a tertiary care environment where prompt thoracotomy and state-of-the-art critical care support are available. For the infant, treatment most often requires a thoracotomy with lobectomy. This can be life saving in critically ill newborns with mediastinal shift and normal lung compression from a ventilated and expanding CCAM. Fig. 61-5 demonstrates the relative size of a right lower lobe CCAM deliberately delivered from the thorax of an infant in extremis. The normal adjacent lung was allowed to ventilate, providing immediate physiologic relief before lobar resection. Because of the long-term risk of infectious complications, surgical resection is considered standard in older patients and in patients without symptoms. It is appropriate in the setting of an acute infectious process to treat a child preoperatively with systemic antibiotics to reduce acute inflammation. Long-term medical management, however, is not appropriate. Approximately 8% of primary lung malignant tumors and 4% of benign tumors are associated with cystic malformation of the lung, including CCAMs (22). To date, at least 24 reports of malignancy occurring within these and other congenital cystic lung lesions add further rationale for surgical resection (22,23,24,25). Pulmonary blastoma and rhabdomyosarcoma are the most common of these malignancies.
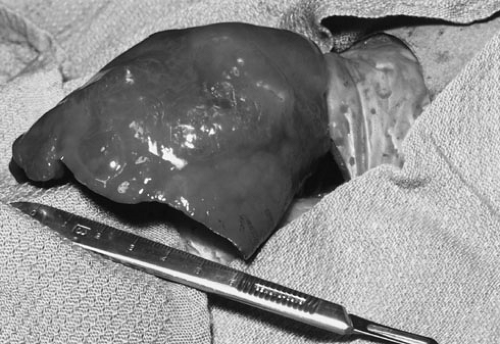 FIGURE 61-5. Right lower lobe cystic adenomatoid malformation that led to acute respiratory distress in a neonate. The size of the lobe after delivery from the thorax is much larger than the volume of the infant thoracic cavity. Delivery of such a space-occupying mass lesion from the thorax can lead to profound and immediate physiologic relief. |
Pneumonectomy is required in as many as 15% to 20% of affected patients to achieve complete resection of a complex or multilobar CCAM (6,7,10). For the limited and well-demarcated CCAM, segmental resection has been reported, but data suggest that operative morbidity may be greater with this approach, and there is little or no apparent long-term benefit.
Outcome after surgical resection of CCAM is generally good. Adzick and colleagues (18) reported survival in four of six selected fetuses with microcystic CCAM after in utero thoracotomy and lobectomy between 24 and 32 weeks’ gestation. For infants who are found at birth to have CCAM, the survival probability with resection is between 80% and 100% in most reports (6,7,10). When it occurs, death is usually the result of respiratory failure in newborns. In older children with infectious presentations, death is rare. The potential complications of neonatal lobectomy for CCAM are not different than for other similar lesions. Although many complications are possible, the overall incidence is less than 10%, and most can be readily managed. Most children have excellent long-term pulmonary function after lobectomy for CCAM. The experience after pneumonectomy is more limited and perhaps less optimistic given the larger extent of the resected lung (see discussion of outcomes after lung resection at the end of this chapter).
Pulmonary Sequestration
Pulmonary sequestration is a form of bronchopulmonary-foregut malformation that gives rise to a mass of lung tissue that is not normally related to the functional lung. In particular, the sequestration can reside outside the lung and be invested with its own visceral pleura (extralobar sequestration), or it can be located within the visceral pleura of the normal lung (intralobar sequestration) (26). The incidence of the two forms is roughly equal. In either case, a sequestration does not communicate with the tracheobronchial tree through a normal bronchus, and the blood supply is derived from one or more anomalous systemic arteries, most often rising from the descending thoracic aorta. In extralobar sequestrations, the arterial blood supply is derived from an infradiaphragmatic source in up to 20% of patients, and venous drainage is typically through the azygous vein. Typically, intralobar sequestrations have venous drainage through the appropriate pulmonary vein. Because of the foregut derivation, occasional
communication with the esophagus or stomach is found. An extralobar sequestration may be located within the diaphragm or even in a subdiaphragmatic position.
communication with the esophagus or stomach is found. An extralobar sequestration may be located within the diaphragm or even in a subdiaphragmatic position.
Many anatomic variations occur with regard to location, the relation to the normal lung, and the systemic arterial blood supply. Conflicting theories of embryogenesis have been offered by way of explanation, distinguishing between disordered budding of the normal tracheobronchial tree and accessory budding from the primitive foregut. The developmental events remain unknown, but the spectrum of bronchopulmonary-foregut malformations is broad. The surgeon who treats these lesions simply must be prepared for variations in the blood supply and relations to the lungs and esophagus. The most important technical concern here is the need to identify the arterial blood supply. Infradiaphragmatic arteries to a pulmonary sequestration are typically elastic and found within the inferior pulmonary ligament. These require precise surgical control to avoid retraction below the diaphragm and occult intraoperative hemorrhage. Communication between the esophagus or stomach and a pulmonary sequestration occurs in about 10% of patients and must be specifically sought, either intraoperatively or possibly preoperatively, by contrast study of the gastrointestinal tract.
Prenatal diagnosis by maternal ultrasound screening is a common mode of discovery of pulmonary sequestrations, particularly for extralobar lesions (27). Typically, a posterior mediastinal or infradiaphragmatic solid mass is observed. The anomalous arterial blood supply may be demonstrable with Doppler ultrasound techniques. Some of these fetuses are vulnerable to the same in utero risks seen with CCAM and congenital diaphragmatic hernia. A large intrathoracic mass lesion can lead to mediastinal compression, hydrops fetalis, and fetal demise. Although relatively uncommon, this is an important possibility to consider in the perinatal care of these fetuses and mothers.
Pulmonary sequestrations are uncommon lesions, but represent about 20% to 40% of the congenital lung bud anomalies in most reports (6,7,10). The male predominance is as high as 3:1 in some reports, particularly for extralobar sequestration. No racial or geographic influence on incidence is known. Newborns have a broad spectrum of presentations, and the lesions can be discovered as asymptomatic posterior mediastinal or abdominal masses. Physiologic derangements include respiratory distress, pneumonia, feeding intolerance, hemorrhage, and congestive heart failure (26). The latter problem results from substantial arteriovenous shunting that can occur within the sequestered lobe, leading to high-output cardiac failure.
Most extralobar sequestrations are diagnosed within the first months or years of life. About 10% to 15% of infants with congenital diaphragmatic hernias have one or more extralobar sequestrations, and this association accounts for their frequent discovery (26). Other congenital anomalies are present in up to 40% of infants and children with extralobar sequestrations, unlike infants with intralobar sequestrations who are otherwise generally normal. Among the defects reported with extralobar sequestrations are ipsilateral diaphragmatic defects, chest wall and vertebral deformities, hindgut duplications, congenital heart disease, and a variety of others (6,7,10).
Intralobar pulmonary sequestrations generally present with infectious sequelae related to inadequate tracheobronchial drainage, either from the lesion or from the adjacent atelectatic lung. Recurrent or persistent pneumonia, lung abscess, and hemoptysis are among the more common presentations. Because these symptoms require temporal evolution, intralobar sequestration has usually presented in childhood or adult life, but not infancy. In published reports, one-half of patients with intralobar sequestrations present after the second decade of life. This pattern of presentation may be changing as prenatal discovery of these lesions by maternal ultrasound has become common (28,29,30).
The imaging evaluation of pulmonary sequestration is relatively straightforward. Prenatal ultrasound discovery is routine and has been mentioned. For newborns with respiratory symptoms, a plain chest radiograph is the standard initial investigation. Ninety percent of extralobar sequestrations appear as posterior mediastinal mass lesions in the left hemithorax. Most commonly, they appear as triangular retrocardiac density on the anteroposterior view (Fig. 61-6). On lateral view, they are posterior to the left
lower lobe at or near the level of the diaphragm. Although many anatomic variations are possible, this appearance is nearly diagnostic on plain radiograph.
lower lobe at or near the level of the diaphragm. Although many anatomic variations are possible, this appearance is nearly diagnostic on plain radiograph.
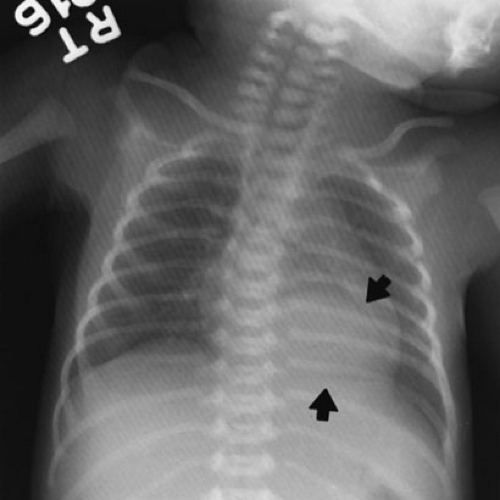 FIGURE 61-6. Chest radiograph of an extralobar pulmonary sequestration (arrows) in the left lower thorax. The contour of the retrocardiac left hemidiaphragm has been obliterated. This was a newborn with no symptoms whose lesion was first noticed on prenatal maternal ultrasound. |
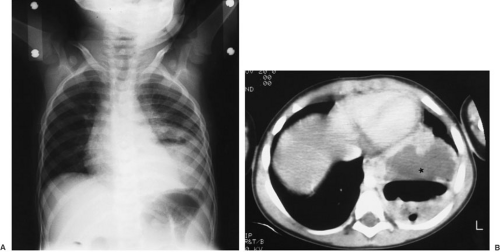 FIGURE 61-7. (A) Plain chest radiograph of a child with fever and cough who has an infected intralobar sequestration of the left lower lobe. (B) Computed tomography scan of the same lesion (asterisk). |
Sixty percent of intralobar sequestrations occur on the left, typically involving the posterior and basal segments of the left lower lobe (6,7,10) (Fig. 61-7). The appearance is generally that of lobar pneumonia and atelectasis. Air–fluid levels may be present if an abnormal communication with the tracheobronchial tree is patent. The intralobar mass, however, may be obscured and the plain films nondiagnostic. Although any lobe can be involved with a sequestration, upper lobe involvement is present in only 10% to 15% of cases, and bilateral sequestrations are rare.
Particularly for intralobar sequestrations, additional imaging is required to establish the diagnosis. Ultrasound, contrast-enhanced CT (see Fig. 61-7B), and MR imaging (Fig. 61-8) all provide excellent anatomic detail, including identification of the systemic vascular blood supply (31). Ultrasound has the potential advantage of Doppler blood flow assessment to identify the arterial supply. Particularly when prenatal ultrasound is the discovery tool, this provides gratifying, correlative data. Angiography, although emphasized in the past, is not required for either diagnosis or operative planning given the modern capabilities of less invasive imaging techniques.
Generally, resection is recommended for both intralobar and extralobar pulmonary sequestrations. Although extralobar sequestrations may be incidental autopsy
findings in the elderly, diagnostic uncertainty about the nature of a posterior mediastinal mass, as well as low but real risks of hemorrhage, infection, and malignancy over a lifetime, must be weighed against the risks of a relatively straightforward surgical resection. Certainly, any symptomatic extralobar pulmonary sequestration requires resection. Simple excision is generally straightforward. By definition, the lesion can be resected without the need to disturb the normal lung. The needs to establish vascular control and rule out foregut communication have been noted.
findings in the elderly, diagnostic uncertainty about the nature of a posterior mediastinal mass, as well as low but real risks of hemorrhage, infection, and malignancy over a lifetime, must be weighed against the risks of a relatively straightforward surgical resection. Certainly, any symptomatic extralobar pulmonary sequestration requires resection. Simple excision is generally straightforward. By definition, the lesion can be resected without the need to disturb the normal lung. The needs to establish vascular control and rule out foregut communication have been noted.
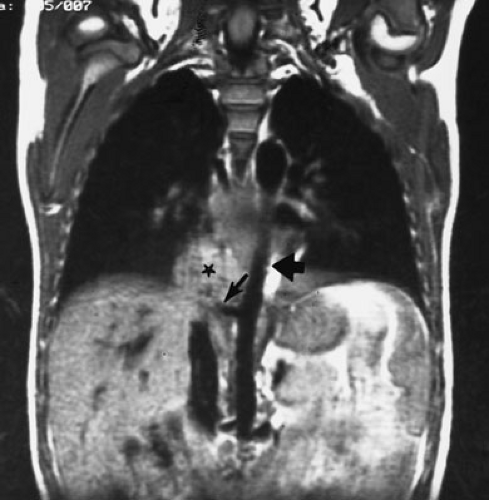 FIGURE 61-8. Magnetic resonance image of a right lower lobe extralobar pulmonary sequestration (asterisk), with the descending aorta visualized (thick arrow) and clear demonstration of the systemic arterial blood supply to the sequestration (thin arrow). (Courtesy of George Bissett, MD, Duke University Medical Center, Durham, NC.) |
Patients with intralobar sequestrations are more likely to present with infection or hemorrhage; therefore, preoperative treatment with systemic antibiotics may be necessary. In the absence of this problem, prompt lobar resection is indicated. The principle for surgical management of an intralobar sequestration is to remove the affected area. In the face of acute or chronic inflammation, this generally requires a formal lobectomy. Although limited segmental resection is feasible in the absence of infection and when the surrounding lung parenchyma is normal, it is practical in only about 25% of patients. Most series report a predominance of lobectomies for intralobar sequestration (9,10). As with extralobar sequestrations, attention must be directed to the systemic arterial blood supply because all the technical issues are similar for intralobar and extralobar lesions. Although standard practice has been thoracotomy and appropriate resection, limited experience with thoracoscopic resection of extralobar sequestrations has been successful, and this approach appears to be appropriate in selected patients.
As with other pulmonary resections in a modern pediatric surgical environment, the survival rate approaches 100% for pulmonary sequestration in the absence of other medical problems. Likewise, the complications and postoperative morbidity are generally minimal. In particular, extralobar resections do not involve removal of normal lung parenchyma, so these children can be expected to have normal pulmonary function and excellent functional outcomes. Reports of intraoperative exsanguination from the loss of control of the systemic arterial blood supply have received considerable attention in the literature historically. This is an important but straightforward technical concern that does not diminish the expectation for an excellent outcome today.
Bronchogenic Cysts and Lung Cysts
A developmental cyst arising from the trachea or a bronchus is referred to as a bronchogenic cyst. These account for about 20% to 30% of congenital bronchopulmonary-foregut cystic malformations (9,10). Potential locations include the cervical or thoracic trachea, the hilar bronchi, or the more distal intraparenchymal bronchi. It has been reported that about 70% of thoracic bronchogenic cysts are located within the lung parenchyma, and the remainder are in the mediastinum, but this distribution varies considerably among different reports (5,6,7,10). Ectopic bronchogenic cysts, including those in paravertebral, paraesophageal, pericardial, subcarinal, and subcutaneous locations, have been reported.
Bronchogenic cysts are typically unilocular mucus-filled lesions arising from the posterior membranous portion of the airway. They do not usually communicate with the functional tracheobronchial tree. Many anatomic variations, however, have been described. By definition, the cyst has structural elements of the airway, including cartilage, smooth muscle, mucous glands, and respiratory epithelium. Likewise, these lesions have a normal bronchial arterial blood supply. The character of the epithelium depends on the site of origin; ciliated columnar, cuboidal, and squamous epithelium are all found within the tracheobronchial tree, and therefore, within these cysts (Fig. 61-9).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


