Liver Physiology and Pathophysiology
Edward P. Tagge
Patrick B. Thomas
Derya U. Tagge
Medical University of South Carolina, Department of Surgery, Divisions of Pediatric Surgery and General Surgery, Charleston, South Carolina 29425.
Medical University of South Carolina, Department of Surgery, Divisions of Pediatric Surgery and General Surgery, Charleston, South Carolina 29425.
Medical University of South Carolina, Department of Surgery, Divisions of Pediatric Surgery and General Surgery, Charleston, South Carolina 29425.
The liver serves a key role in many critical metabolic pathways. As the first organ to receive a nutrient-enriched blood supply from the portal system, it is strategically situated to perform a large number of diverse metabolic functions. In addition, the unique vascular structure of the liver provides unparalleled access to nutrients and xenobiotics absorbed from the intestinal lumen. Processing, redistribution, and storage of metabolic fuels such as glucose and fatty acids are a major responsibility of the liver. Protein production, particularly albumin and the coagulation factors, is an extremely important function of the liver. The liver also contains a host of biochemical pathways for the modification and detoxification of compounds absorbed from the small intestine.
ANATOMY
Morphogenesis
The liver and the biliary system originate from the primitive foregut. The hepatic anlage appears during the fourth week of gestation as a duodenal diverticulum. The hepatic lobules are identifiable at the sixth gestational week, and the cystic duct and the gallbladder are fully recanalized by the seventh to eighth week. The liver reaches a peak relative size of about 10% of fetal weight at the ninth week, dropping to 5% of body weight at birth and eventually 2% in an adult. Early in gestation, hematopoietic cells outnumber functioning hepatocytes in the hepatic anlage. Near term, the hepatocytes dominate the organ, and hematopoiesis is virtually absent by the second postnatal month in full-term infants.
Fetal hepatic blood flow is derived from the hepatic artery and from the portal and umbilical veins, which form the portal sinus. The portal venous inflow is directed mainly to the right lobe of the liver; and umbilical flow is directed primarily to the left. The ductus venosus shunts blood from the portal and umbilical veins to the hepatic vein, bypassing the sinusoidal network. Because the oxygen saturation is lower in portal than in umbilical venous blood, the right hepatic lobe has lower oxygenation and greater hematopoietic activity than the left hepatic lobe. The ductus venosus obliterates when oral feedings are initiated, ensuring all blood traverse the hepatic vascular sinusoidal system.
Ultrastructure
The fundamental unit in the liver is the hepatic acinus, which can be envisioned as a wheel, with the central vein comprising the hub and four to six portal triads representing the rim. The transport and metabolic activities of the liver are facilitated by the structural arrangement of liver cell cords, which are formed by rows of hepatocytes, separated by sinusoids that converge toward the tributaries of the central vein. This relationship establishes the patterns of flow for substances to and from the liver. Plasma proteins and other plasma components are secreted by the liver. Absorbed and circulating nutrients arrive through the portal vein or the hepatic artery, pass through the sinusoids where they are modified by the hepatocytes, and are
returned to the systemic circulation at the central vein. Biliary components are transported via the series of enlarging channels from the bile canaliculi through the bile ductule to the common bile duct.
returned to the systemic circulation at the central vein. Biliary components are transported via the series of enlarging channels from the bile canaliculi through the bile ductule to the common bile duct.
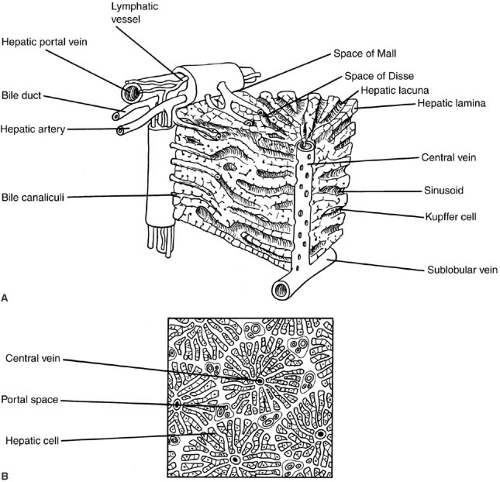 FIGURE 90-1. Liver lobule. (Adapted from Anatomical Charts, Co.) |
Our understanding of the ultrastructural anatomy of the hepatocyte has been made possible through electron microscopy and cell fractionation techniques (Fig. 90-1). Hepatocytes are epithelial cells that are bounded by three separate membrane domains: (1) the basolateral membrane, which abuts the sinusoidal space; (2) the apical membrane, which circumscribes the canaliculus; and (3) the lateral hepatic membrane between adjacent hepatocytes (1). The liver’s unique sinusoidal structure is well suited for the bidirectional transfer of a variety of solutes, including macromolecules, across the sinusoidal membrane. The low pressure allows blood to percolate slowly through the sinusoids and hepatic acinus. Fenestrae within the sinusoidal endothelium and the absence of a basement membrane permit direct contact of the portal blood with the hepatic sinusoidal surface in the space of Disse. Canalicular membranes of adjacent hepatocytes form bile canaliculi, which are bounded by tight junctions preventing transfer of secreted compounds back into the sinusoid. There are abundant mitochondria, which are the sites of substrate metabolism, of key processes in gluconeogenesis, and of storage and release of energy. An extensive endoplasmic reticulum is the site of protein and triglyceride synthesis and drug metabolism. The prominent golgi apparatus is active in protein packaging and possibly in bile secretion.
There are a variety of nonparenchymal cells in the liver, which serve other important physiologic roles. The sinusoidal space houses macrophage-derived Kupffer cells, which are phagocytic cells. These cells are important mediators of the hepatic inflammatory response. The lipocyte, identified phenotypically by its high lipid content, is the major site for vitamin A storage. It is also the major cell type responsible for synthesis of extracellular collagen, and is a critical component of the fibrogenic response to liver injury.
METABOLIC FUNCTIONS
A major responsibility of the liver is to provide a continual source of energy for the body. The liver regulates nutrient flux during periods of nutrient absorption, digestion, and modification for storage. It also makes nutrients available systemically during periods of fasting. The regulation of these metabolic pathways involves complex interactions among the nutrient content of the blood, end products of nutrient metabolism, and hormonal regulation.
Carbohydrate Metabolism
Glucose is the central component of all metabolic pathways because it can be converted to amino acids, fatty acids, or glycogen (the major storage form of glucose). Maintaining adequate circulating levels of glucose is essential for the central nervous system. The brain normally uses glucose as its major metabolic fuel, and hepatic glycogen is the main storage site for the glucose-dependent brain, erythrocytes, retina, and renal medulla (2). The liver serves a key role in maintaining total carbohydrate stores because of its ability to store glycogen and synthesize glucose from precursors (3). Glycogen constitutes approximately 10% of liver weight. The liver can store approximately a 2-day supply of glucose in the form of glycogen before gluconeogenesis occurs. By gluconeogenesis, the liver is able to produce up to 240 g of glucose a day, which is approximately twice the metabolic needs of the retina, red blood cells, and brain.
Immediately after birth, an infant is dependent on hepatic glycogenolysis; thereafter, an infant is capable of both glycogenolysis and gluconeogenesis. The fluctuations in serum glucose concentration in preterm infants are due in part to the fact that efficient regulation of the synthesis, storage, and degradation of glycogen develops only near the end of full-term gestation.
Fatty Acids Metabolism
The liver plays a central role in regulating the body’s total fatty acid needs. Oxidation of fatty acids to carbon dioxide and water yields the highest adenosine triphosphate (ATP) production of any metabolic fuel. Fatty acids are thus the most efficient long-term storage form of energy. Fatty acid oxidation particularly provides a major source of energy in early life, complementing glycogenolysis and gluconeogenesis. Newborn infants are relatively intolerant of prolonged fasting, owing in part to a restricted capacity for hepatic ketogenesis. Rapid maturation of the ability of the liver to oxidize fatty acid occurs during the first few days of life. Because milk provides the major source of calories in early life, this high-fat, low-carbohydrate diet mandates active gluconeogenesis to maintain blood sugar levels. When the glucose supply is limited, ketone body production from endogenous fatty acids provides substrate for hepatic gluconeogenesis and an alternative fuel for brain metabolism. Excess glucose can be converted to fatty acid for future use and stored in distal sites such as adipose tissue.
Fatty acids are also involved in various physiologic functions. For instance, fatty acids are constituent parts of various cellular structural components such as membranes. Furthermore, they are involved in regulatory functions in intracellular communication, golgi function, and anchoring of membrane proteins. The liver serves a central role in the synthesis of fatty acid for storage in distal sites and the trafficking of lipids within the body. For lipids to be transferred between different locations, the liver synthesizes a large number of apolipoproteins.
Lipids may be stored temporarily in the liver as fat droplets, excreted directly into bile, or metabolized into bile acids. The liver is the major site of sterol excretion from the body and is the site of bile acid synthesis. The production and metabolism of sterols require complex regulation of multiple enzymatic pathways, and bile acids play a critical role in modulating these enzyme activities. Bile acids are recirculated 20 to 30 times per day via a unique enterohepatic circulation that requires specific active transport processes (4) (Fig. 90-2). In the terminal ileum, a unique sodium-dependent bile acid transporter prevents loss of bile acids in the stool (5), and in the liver the activity of specific transmembrane transporters and intracellular binding proteins are required to capture and recirculate the bile acids. Bile acids also play an important role in micellization of fats for intestinal absorption.
Protein Metabolism
Formation of plasma proteins is a vital function of the liver (Table 90-1). Because protein metabolism is such an important part of liver function, the synthetic capability of the liver is routinely assessed by measurement of specific serum proteins it produces. The liver synthesizes blood coagulation proteins factor II (prothrombin), factor VII, factor IX, factor X, protein C, and protein S. All these proteins undergo a unique, vitamin K-dependent gamma-carboxylation of specific glutamic acid residues, which is essential for normal activity.
More than 95% of serum α1-antitrypsin (α1-AT), a member of the serine protease inhibitor family, is synthesized by the liver. Absent or reduced α1-AT activity is manifested clinically by destruction of the lung parenchyma with the early onset of emphysema. The liver also synthesizes carrier proteins albumin and alpha-fetoprotein (AFP). Albumin functions as a nonspecific carrier protein that binds fatty acids, bile acids, and numerous other compounds, as well as providing serum oncotic pressure that opposes hydrostatic pressure. AFP serves a similar function in the
developing organism. Synthesis of albumin appears at approximately the seventh and eighth gestational week and increases in inverse proportion to that of AFP, which is a dominant fetal protein. By the third and fourth month of gestation, the fetal liver is able to produce each of the major protein classes, at concentrations considerably below those achieved at maturity. AFP is entirely replaced by serum albumin at the end of the first year of life. During liver regeneration, as in acute viral hepatitis, and in liver tumors such as hepatoblastoma and hepatocellular carcinoma, serum AFP levels may be elevated.
developing organism. Synthesis of albumin appears at approximately the seventh and eighth gestational week and increases in inverse proportion to that of AFP, which is a dominant fetal protein. By the third and fourth month of gestation, the fetal liver is able to produce each of the major protein classes, at concentrations considerably below those achieved at maturity. AFP is entirely replaced by serum albumin at the end of the first year of life. During liver regeneration, as in acute viral hepatitis, and in liver tumors such as hepatoblastoma and hepatocellular carcinoma, serum AFP levels may be elevated.
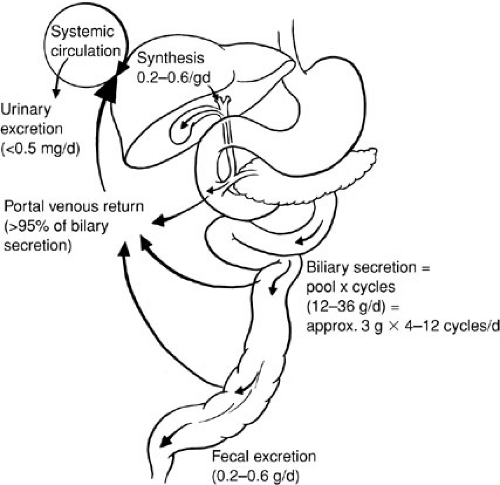 FIGURE 90-2. Enterohepatic circulation of bile acids. (From Carey MC. The enterohepatic circulation. In: Arias IM, Popper H, Schacter D, et al., ed. The liver: biology and pathobiology. New York: Raven Press, 1982:430, with permission.) |
TABLE 90-1 Important Proteins Produced by the Liver. | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Biotransformation
The liver plays a major role in the biotransformation of drugs, particularly those poorly excreted in the urine. This occurs in two phases. Phase I uses microsomal enzymes such as the cytochrome P450 enzymes to make the drugs more polar. In phase II, the drugs are conjugated with other moieties to facilitate their excretion in bile or urine. Newborn infants have a decreased capacity to metabolize metabolic byproducts and detoxify certain drugs, owing to underdevelopment of these hepatic enzyme systems. For example, newborn infants have decreased activity of uridine diphosphate (UDP)-glucuronyl transferase (rate-limiting enzyme in bilirubin excretion), which converts unconjugated bilirubin to the readily excreted glucuronide conjugate. Microsomal activity can be stimulated by the administration of phenobarbital or other inducers of cytochrome P450.
Hepatic Excretory Function
Bile acids are the major product of degradation of cholesterol, and their secretion is the major determinant of bile flow. Incorporation of bile acids into micelles with cholesterol and phospholipid creates an efficient vehicle for solubilization and intestinal absorption of lipophilic compounds, such as dietary fats and fat-soluble vitamins. The primary bile acids, cholic acid and chenodeoxycholic acid, are synthesized in the liver. In response to a meal, contraction of the gallbladder delivers bile acids to the intestine. After mediating fat digestion, the bile acids enter the enterohepatic recirculation as described previously. Bile acids are reabsorbed from the terminal ileum and returned to the liver via portal blood, where they are taken up by liver cells and reexcreted in bile. In an adult, this enterohepatic circulation involves 90% to 95% of the circulating bile acid pool. Bile acids that escape ileal reabsorption reach the colon, where the bacterial flora, through dehydroxylation and deconjugation, produces the secondary bile acids, deoxycholate and lithocholate.
Neonates have inefficient ileal reabsorption and a low rate of hepatic clearance of bile acids from portal blood. The latter results in elevated serum concentrations of bile acids in healthy newborns, often to levels that would suggest liver disease in older individuals. The size of the bile acid pool in a neonate is about one-half that of an adult, and the bile acid concentration in the proximal intestinal lumen is similarly decreased to levels that are frequently below the concentration required for micelle formation (2 mM); accordingly, absorption of dietary fats and fat-soluble vitamins is reduced, but not sufficiently to produce malabsorption. Transient phases of “physiologic cholestasis” and “physiologic steatorrhea” have a role in the nutrition of low birth weight infants, but are otherwise of minor importance.
CLINICAL MANIFESTATIONS OF LIVER DISEASE
Acute or chronic liver injury can reduce the metabolic and synthetic capabilities of the liver, thereby resulting in diverse clinical disorders and a variety of clinical presentations. Liver enlargement, although not pathognomonic, is one of the more common presentations of liver disease. In a newborn infant, extension of the liver edge more than 3.5 cm below the costal margin in the midclavicular line suggests hepatic enlargement. In children, the normal liver edge can be felt up to 2 cm below the right costal margin. The downward displacement of the liver by the diaphragm or thoracic organs can create an erroneous impression of hepatomegaly. Examination of the liver should note the consistency, contour, tenderness, or the presence of any masses or bruits, as well as assessing splenic size. Ultrasonography can often help in evaluating unexplained hepatomegaly.
Jaundice becomes clinically apparent in children and adults when the serum concentration of bilirubin reaches 2 to 3 mg per dL. Bilirubin occurs in four forms: (1) unconjugated bilirubin tightly bound to albumin, (2) free or unbound bilirubin (the form responsible for kernicterus), (3) conjugated bilirubin, and (4) delta fraction (bilirubin covalently bound to albumin). Jaundice may reflect accumulation of either unconjugated or conjugated bilirubin. An increase in unconjugated bilirubin may indicate increased production, hemolysis, reduced hepatic removal, or altered metabolism of bilirubin. Significant accumulations of conjugated bilirubin (more than 20% of total) reflect decreased excretion by damaged hepatic parenchymal cells or disease of biliary tract. Cholestasis is a more general term, defined as accumulation in serum of substances normally excreted in bile, such as bilirubin, cholesterol, bile acids, and trace elements.
Cirrhosis is an advanced form of liver disease, and may result from hepatitis, toxic injury, or chronic biliary obstruction. Cirrhosis is defined histologically by the presence of bands of fibrous tissue linking portal areas, thus forming parenchymal nodules. Cirrhosis may be macronodular, with nodules of various sizes (up to 5 cm)
separated by broad septa, or micronodular, with nodules of uniform size (less than 1 cm) separated by fine septa. The progressive scarring of cirrhosis leads to altered hepatic blood flow, further impairing liver cell function and causing the development of portal hypertension (PH).
separated by broad septa, or micronodular, with nodules of uniform size (less than 1 cm) separated by fine septa. The progressive scarring of cirrhosis leads to altered hepatic blood flow, further impairing liver cell function and causing the development of portal hypertension (PH).
PH is defined as an increase in portal venous pressure to greater than 20 mm Hg. Because the portal vein drains the splanchnic area into the hepatic sinusoids, pressure is normally slightly higher (5 to 10 mm Hg) in the portal vein than in other venous systems in order to overcome the resistance of the sinusoidal system. However, in liver disease, a combination of increased hepatic resistance and increased circulating blood volume can lead to hypertension in the portal system, manifesting as gastrointestinal (GI) bleeding at points of portosystemic collaterals.
Ascites is a common manifestation of end-stage liver disease. In patients with significant hepatic disease, sinusoidal blockade caused by cirrhosis increases hydrostatic pressure and transudation of fluid.
Metabolic abnormalities may complicate acute or chronic liver disorders, leading to encephalopathy. This neuropsychiatric disturbance can present in a variety of ways, including restless, altered mentation, varying levels of consciousness, or even coma. With chronic liver disease, hepatic encephalopathy may be recurrent, precipitated by bleeding, infection, drugs or electrolyte and acid–base disturbances.
There is a close relationship between hepatic and renal dysfunction. Systemic disease may affect both organs individually and/or simultaneously, or parenchymal liver disease may produce secondary impairment of renal function. Hepatorenal syndrome is defined as renal failure in a patient with cirrhosis and no other demonstrable cause of renal failure. The pathophysiology is poorly defined, but seems to involve altered hormonal metabolism that leads to abnormalities of renal blood flow.
EVALUATION OF LIVER DISEASE
Adequate evaluation of a child with suspected liver disease starts with an accurate history and a careful physical examination. Further evaluation is aided by judicious selection of diagnostic tests, followed by imaging studies and potentially a liver biopsy. Any single biochemical assay provides limited information, which must be placed in the context of the entire clinical picture.
Biochemical Liver Tests
The liver performs a diverse array of biochemical, synthetic, and excretory functions, and as a result, no single biochemical test is capable of providing an accurate assessment of hepatic function. Biochemical liver tests have limited sensitivity and specificity, and do not all reflect liver function as the common misnomer “liver function tests” implies (6).
The aminotransferases include aspartate aminotransferase (AST, formerly SGOT) and alanine aminotransferase (ALT, formerly SGPT). These enzymes are elevated in many forms of liver disease, especially those that are associated with significant hepatocyte necrosis, such as seen in ischemic injury. Although ALT is relatively liver specific, AST is also found in skeletal and cardiac muscle, kidney, brain, pancreas, and blood cells. Lactate dehydrogenase (LDH) is a very nonspecific assay, as elevated levels are seen with skeletal or cardiac muscle injury, hemolysis, stroke, and renal infarction, in addition to acute and chronic liver disease.
Alkaline phosphatase (AP) comprises a group of enzymes present in a large variety of tissues, particularly bone and liver. Elevation of AP in the setting of liver disease results from increased synthesis and from release of the enzyme into serum, rather than from impaired biliary secretion. Levels of AP up to three times normal are relatively nonspecific. Because marked elevations of AP can be seen with both infiltrative hepatic disorders and biliary obstruction (intra- or extrahepatic), the level of AP cannot be used to distinguish between them. Elevation of AP is commonly seen in neonatal liver disease of various causes. Hepatic gamma glutamyl transpeptidase (GGTP) is derived from hepatocytes and biliary epithelia. Like AP, GGTP is found in the liver and many extrahepatic tissues, including the kidney and pancreas. However, it is not found in appreciable quantities in bone, and it is thus helpful in confirming the hepatic origin of an elevated AP level. The normal serum GGTP level is significantly higher in infants than in adults. Benign recurrent intrahepatic cholestasis and Byler’s syndrome, both rare cholestatic liver diseases that often present in infancy, are characterized by elevation of the serum AP without an elevated GGTP.
Bilirubin is an organic anion that is derived primarily from the catabolism of hemoglobin. Serum bilirubin consists of two major forms, a water-soluble, conjugated, “direct” fraction and a lipid-soluble, unconjugated, “indirect” fraction. The serum bilirubin level is normally almost entirely unconjugated, reflecting a balance between the rates of production and hepatobiliary excretion. Unconjugated hyperbilirubinemia (i.e., indirect bilirubin fraction greater than 85% of the total serum bilirubin) results from either increased bilirubin production (most likely by hemolysis) or from defects in hepatic uptake or conjugation. Conjugated hyperbilirubinemia (i.e., direct bilirubin fraction greater than 50% of the total serum bilirubin) occurs as a result of a defect in hepatic excretion, with subsequent regurgitation of conjugated bilirubin from hepatocytes into the serum. This impaired biliary excretion occurs in both parenchymal liver disease and biliary tract obstruction (7). Thus, measurement of the conjugated fraction does not reliably distinguish biliary obstruction from parenchymal
liver disease. Because conjugated bilirubin is cleared by the kidney, serum concentrations of bilirubin rarely exceed 30 mg per dL in the absence of hemolysis or renal failure.
liver disease. Because conjugated bilirubin is cleared by the kidney, serum concentrations of bilirubin rarely exceed 30 mg per dL in the absence of hemolysis or renal failure.
The liver plays a crucial role in hemostasis. All major coagulation factors except factor VIII are synthesized in hepatocytes (see Chapter 16, Fig. 16-2) The prothrombin time (PT) measures the rate of conversion of prothrombin to thrombin and reflects the activity of several of the factors involved in the extrinsic coagulation pathway, including factors II, V, VII, and X. Vitamin K is required for the gamma-carboxylation of factors II, VII, IX, and X, which is essential for the normal function of these factors. Prolongation of the PT may occur in decompensated liver disease with hepatocellular dysfunction and in chronic cholestatic disease with fat malabsorption and concomitant vitamin K deficiency.
Approximately 10 g of albumin is synthesized and secreted by hepatocytes each day. With progressive parenchymal liver disease, albumin synthetic capacity decreases. Thus, albumin concentrations are believed to reflect one of the important synthetic functions of the liver. However, the serum albumin concentration reflects a variety of extrahepatic factors, including nutritional and volume status, vascular integrity, catabolism, hormonal factors, and loss in the urine or stool. Therefore, a low serum albumin level is not specific for liver disease.
Liver Biopsy
Because the morphologic features of specific hepatic diseases are frequently distinctive, liver biopsy combined with clinical data can suggest an etiology. Tissue obtained by percutaneous liver biopsy can be used for histologic examination, for enzyme analysis to detect inborn errors of metabolism, and for analysis of stored material (e.g., iron, copper). In infants and children, needle biopsy of the liver is easily accomplished through the percutaneous approach. The procedure can be performed safely in infants as young as 1 week, and patients usually require only sedation and local anesthesia. The risk of development of a complication such as hemorrhage, hematoma, creation of an arteriovenous fistula, pneumothorax, or bile peritonitis is very small. In patients with significant liver disease, correction of any associated coagulopathy is strongly encouraged prior to percutaneous liver biopsy. If the coagulopathy is not correctable, liver biopsy under direct visualization is a relatively straightforward procedure.
Imaging Procedures
Various techniques help define the size, shape, and architecture of the liver, as well as the anatomy of the biliary system. A plain roentgenographic study may suggest hepatomegaly. Calcifications or collections of gas may be evident within the liver, biliary tract, or portal circulation.
Ultrasonography provides extremely useful information about the size, composition, and blood flow of the liver. Ultrasound is particularly useful in children because it can be done at the bedside without the need for sedation or general anesthesia. Even in neonates, ultrasonography can assess gallbladder size, visualize gallstones, detect dilatation of the biliary tract, and define a choledochal cyst. In patients with PH, ultrasonography can evaluate patency of the portal vein or demonstrate collateral circulation. Small amounts of ascitic fluid can be easily seen on ultrasound, as can mass lesions as small as 1 to 2 cm.
Computed tomography (CT) scanning provides more detailed information, but is less suitable for use in younger patients because of the small size of structures and the paucity of intraabdominal fat for contrast. When a hepatic tumor is suspected, CT scanning is the best method to define anatomic extent, solid or cystic nature, and vascularity. Either CT scanning or ultrasonography may be used to guide percutaneously placed fine needles for biopsies or aspiration of specific lesions.
Occasionally, direct visualization of the intrahepatic and extrahepatic biliary tree may be required in some patients to evaluate the cause, location, or extent of biliary obstruction. Gastroduodenoscopy with endoscopic retrograde cholangiopancreatography (ERCP) has been performed in children for more than 20 years (8). The technique has been quite successful in managing children with a large number of biliary disorders (9,10). More recently ERCP has even been used in some centers to delineate the extrahepatic ductal anatomy in infants, and has been successful in making a differential diagnosis of neonatal hepatitis from extrahepatic biliary atresia (BA) (11,12). Occasionally, percutaneous transhepatic cholangiography has been used to outline the biliary ductal system in infants and young children. Magnetic resonance imaging (MRI) is receiving increasing attention as an alternative imaging technology; magnetic resonance cholangiopancreatography can be of value in differentiating biliary tract lesions (13).
Radionuclide scanning relies on selective uptake of a radiopharmaceutical agent. Commonly used agents include (1) technetium 99m-labeled sulfur colloid, which undergoes phagocytosis by Kupffer cells and (2) 99m Tc-iminodiacetic acid agents, which are taken up by hepatocytes and excreted into bile. The 99m Tc-sulfur colloid scan may detect focal lesions (e.g., tumors, cysts, or abscesses) greater than 2 to 3 cm in diameter. The 99mTc-substituted iminodiacetic acid dyes may differentiate intrahepatic cholestasis from extrahepatic obstruction in neonates. Imaging results are best when scanning is preceded by a 5- to 7-day period of treatment with phenobarbital to stimulate bile flow.
Selective angiography of the celiac, superior mesenteric, or hepatic artery may be used to visualize the hepatic or portal circulation. Angiography is occasionally used to
define the blood supply of tumors before surgery and is useful in the study of patients with known or presumed PH. The patency of the portal system, the extent of collateral circulation, and the caliber of vessels under consideration for a shunting procedure can be evaluated. MRI and variations on CT scanning can now provide similar information.
define the blood supply of tumors before surgery and is useful in the study of patients with known or presumed PH. The patency of the portal system, the extent of collateral circulation, and the caliber of vessels under consideration for a shunting procedure can be evaluated. MRI and variations on CT scanning can now provide similar information.
CHOLESTASIS
Neonatal Cholestasis
Neonatal cholestasis must always be considered in a newborn who is jaundiced for more than 14 to 21 days; measurement of the serum total and conjugated bilirubin in these infants is mandatory (14). Conjugated hyperbilirubinemia, dark urine, and pale stools are pathognomonic of the neonatal hepatitis syndrome that should be investigated urgently. This neonatal hepatitis syndrome has many causes—infectious, genetic, metabolic, or miscellaneous. These abnormalities give rise either to mechanical obstruction of bile flow or to functional impairment of hepatic excretory function and bile secretion (Table 90-2). Neonatal cholestasis may be divided into extrahepatic and intrahepatic disease. The most important condition in the differential diagnosis is BA because these infants require expeditious surgical drainage. However, differentiation among extrahepatic BA, idiopathic neonatal hepatitis, and intrahepatic cholestasis is often particularly difficult.
TABLE 90-2 Causes of Neonatal Cholestasis Syndrome. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The clinical features of infants with neonatal cholestasis provide very few clues about etiology (15). The initial step is prompt recognition of any specific or treatable primary causes of cholestasis, such as sepsis, an endocrinopathy (hypothyroidism or panhypopituitarism), nutritional hepatotoxicity caused by a specific metabolic illness (galactosemia), or other metabolic diseases (tyrosinemia). Recognition of such entities allows institution of appropriate therapy and may possibly prevent further injury. Hepatobiliary disease may be the initial manifestation of homozygous α1-AT deficiency or of cystic fibrosis. Neonatal liver disease may also be associated with congenital syphilis and specific viral infections, notably echo virus, herpes virus, and the hepatitis viruses. Additional cholestatic disorders include neonatal iron storage disease and inborn errors of bile acid biosynthesis.
BA is a disorder of the infant liver in which there is obliteration or discontinuity of the extrahepatic biliary system, resulting in obstruction of bile flow. Untreated, the resulting cholestasis leads to progressive conjugated hyperbilirubinemia, cirrhosis, hepatic failure, and subsequent death within 2 years. The natural history of BA has been favorably altered by the Kasai portoenterostomy; approximately one-fourth of patients who undergo a Kasai portoenterostomy will survive more than 10 years without liver transplantation, one-fourth drain bile but develop cirrhosis, and the remaining one-half of patients never experience adequate bile flow (16). The portoenterostomy should be performed before there is irreversible sclerosis of the intrahepatic bile ducts; this is before the child reaches 3 months of age (17). BA represents the most common indication for pediatric liver transplantation (18,19,20).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


