Key Principles in Human Genetics
Michael J. Bamshad and Lynn B. Jorde
Genetic disorders and birth defects are sometimes perceived as being so uncommon that the general pediatrician will seldom encounter them. However, each day more than 400 babies with birth defects are born in the United States, and 1 of every 5 infant deaths is caused by these disorders. Virtually every medical condition, except for some cases of trauma, is influenced by an individual’s genetic background. Rapid advances in our knowledge of the genetic basis of disease, coupled with emerging technologies for high-throughput genetic testing, are also rapidly transforming the practice of medicine.
Comprehensive databases of factual information about the identification of new disease genes, genotype–phenotype correlation, and the availability of genetic testing have become widely available online and in published review articles. Thus, each of the following sections seeks to emphasize important fundamental concepts while providing paradigmatic examples.
Traditionally, various human traits (eg, skin color, intelligence) and disorders (eg, phenylketonuria, achondroplasia) have been divided into those that are genetic versus those that are nongenetic, the latter usually referring to disorders “determined” by the environment (eg, infectious diseases, teratogens). However, this dichotomy separating gene and environment is largely artificial, and the distinction between the two has become increasingly blurred. Disorders caused by mutations in a single gene (eg, cystic fibrosis, sickle-cell disease) can be heavily influenced by the environment, and diseases caused by the environment (eg, AIDS secondary to infection with HIV-1) can be substantially modified by the presence of certain polymorphisms. In other words, the cause of most diseases can be considered genetic and environmental; some diseases are more strongly influenced by genes, whereas others are more strongly influenced by the environment.
Whether defined narrowly or broadly, genetic disorders and birth defects contribute substantially to morbidity and mortality in pediatric patients. For example, the percentage of deaths attributable to genetic disease in hospitals in the United Kingdom has risen from 16.5% in 1914 to 50% in 1976. Birth defects are also the leading cause of death in infancy in the United States, accounting for more than 20% of infant deaths. Cardiovascular malformations are the leading cause of premature mortality from congenital anomalies. This reflects a better understanding of the etiology of pediatric diseases as well as the substantial reduction in infectious disease and perinatal mortality during the 20th century. Population-based studies suggest that birth defects and genetic disorders account for about 10% of hospitalizations and about 30% of all hospitalization charges. Approximately 7% of pediatric admissions are for single-gene and chromosomal disorders, and another 15% to 20% for congenital malformations of different types. Moreover, about 35% of hospital deaths of children are caused by a genetic condition and/or birth defect. Recent advances in therapeutics have substantially prolonged the survival of children with birth defects and/or genetic diseases, challenging practitioners to care for older children and young adults with genetic disorders.
CLASSIFICATION OF GENETIC DISORDERS
Classifications of genetic disorders depend, in part, on the delineation of a phenotypic trait (eg, mental retardation, craniosynostosis) and the judgment of whether this trait varies substantially from a “normal” trait. Whether a trait should be considered normal or abnormal (eg, a midline abdominal aorta, short digits) is not always clear, and the decision may depend, in part, on findings in other family members. Genetic disorders can be broadly classified into several major groups: chromosome disorders, single-gene disorders, multifactorial disorders, and disorders with nontraditional mechanisms of expression and inheritance.
Chromosome disorders result from abnormalities of chromosome number (ie, aneuploidy) and/or structure. These abnormalities include duplications, deletions, rearrangements, extra chromosomes (trisomy), and missing chromosomes (monosomy). Most chromosome disorders arise de novo and are not transmissible. However, some chromosome disorders are heritable because they are carried by an unaffected parent in a state that does not cause disease (eg, balanced rearrangements) but can be transmitted to an offspring to produce a state that does cause disease (unbalanced rearrangement). Most conceptions with chromosome abnormalities are terminated spontaneously early in gestation, and chromosome abnormalities are the leading known cause of pregnancy loss. Most individuals with chromosome disorders have major and/or minor malformations, variable degrees of mental retardation, and/or growth deficiency.
Disorders in which single genes have been altered are called monogenic conditions. Single-gene disorders can be transmitted from parent to offspring in autosomal-dominant or autosomal-recessive patterns and thus are sometimes known as mendelian conditions. Single-gene disorders can also be produced by new mutations in a single individual that are appearing for the first time in a family; the mutation has arisen de novo in the affected family member. Copies of genes that have different sequences are referred to as alleles. A gene’s location on a chromosome is termed a locus.
The four modes of inheritance of single-gene disorders are autosomal-recessive, in which both copies of a gene at a single locus on an autosome (nonsex chromosome) are altered; autosomal-dominant, in which only one allele of a gene at a single locus on an autosome is altered; X-linked recessive, in which one allele of a gene at a locus on the X chromosome is altered in affected males and both alleles of a gene at a locus on the X chromosome are altered in affected females; and X-linked dominant, in which one allele of a gene at a locus on the X chromosome is altered in affected males and females. Y-linked inheritance is uncommon for genetic disorders.
Substantial progress has been made toward identifying the genes that cause single-gene disorders. However, these conditions represent only a small fraction of the total burden of genetic disease. Most birth defects are not caused by alterations of a single gene or chromosome, and many common pediatric disorders are influenced by different combinations of alleles at various loci. Traits in which variation is caused by the effects of many different genes are called polygenic. If the variation of a polygenic trait is also affected by nongenetic factors (eg, environmental variables), the terms multifactorial or complex are often used to describe the trait. In contrast to single-gene disorders, multifactorial traits are not transmitted in mendelian patterns. The identification of the genetic and environmental determinants of multifactorial traits is a major objective of genetic research.
Some genetic disorders do not segregate in the patterns expected of single-gene conditions or multifactorial traits and are transmitted from parent to offspring in nontraditional patterns of inheritance. Disorders exhibiting nontraditional patterns of inheritance include those caused by mutations in the mitochondrial genome, uniparental disomy (two alleles from the same locus inherited from one parent instead of both parents), and gene duplication. The expression of some conditions is influenced substantially by whether the altered allele is transmitted to a child by the mother or father, a so-called parent-of-origin effect. Some genetic conditions exhibit a nontraditional pattern of inheritance as a consequence of this parent-of-origin effect and are relatively uncommon.
KEY PRINCIPLES IN HUMAN GENETICS
The terms congenital, hereditary, and familial are not synonymous. Conditions that are present at birth are referred to as congenital conditions, whether the major risk factor is genetic or not. For example, infection with cytomegalovirus, clubfeet caused by oligohydramnios, and trisomy 21 are all congenital conditions. However, only trisomy 21 is clearly a genetic condition, and none of these conditions are usually hereditary.
The term hereditary defines conditions that can be transmitted from parent to offspring. All hereditary conditions are genetic conditions, but not all genetic conditions are hereditary (eg, most cases of trisomy 21). Conditions that appear to cluster within families are frequently called familial conditions and may include non-genetic conditions (eg, rotaviral or streptococcal infections) as well as genetic conditions. Nevertheless, although all hereditary conditions can be familial, not all genetic conditions are familial (eg, most cases of trisomy 21). Distinguishing which term most appropriately defines a condition facilitates diagnosis, management, and counseling of families about the risk of recurrence of a condition.
The probability that an individual who possesses a disease-related genotype (an individual’s allelic constitution at a single locus is called a genotype) exhibits the disease phenotype is called penetrance. When this probability is less than 1, the disease is said to exhibit reduced (or incomplete) penetrance. Penetrance levels are usually estimated by examining a large number of families and determining what proportion of the obligate carriers (ie, those individuals who have an affected parent and an affected child and thus must be carriers of the altered gene) or obligate homozygotes (in the case of autosomal-recessive disorders) develop the disease phenotype. Retinoblastoma is a good example of a genetic condition in which reduced penetrance is observed. Family studies have demonstrated that about 10% of obligate carriers of a mutation in the retinoblastoma susceptibility gene do not develop a retinoblastoma. The penetrance of the condition is thus about 90%.
Individuals with the same genetic condition or even the same genotype can have substantially different phenotypes. This is called variable expressivity. For example, within the same kindred, one sibling with neurofibromatosis type 1 (NF1) may have café au lait spots, axillary freckling, and sphenoid wing dysplasia, and another sibling with NF1 has café au lait spots, a plexiform neurofibroma, and pseudarthrosis of the tibia. Although each sibling has the same genotype, the expression of the NF1 phenotype is different between them.
Variable expressivity may be explained by the influence of other genes (ie, modifying genes), environmental factors (eg, earlier palliative intervention), or random variation. Variable expressivity among families is sometimes related to the presence of different genotypes. The causation of a disease phenotype by a variety of different genotypes at the same locus is called allelic heterogeneity. A potentially powerful strategy to provide affected individuals with better anticipatory guidance is to estimate the correlation between genotypes and specific phenotypic characteristics (genotype-pheno-type correlation studies). The compilation of findings from many individuals with a genetic condition defines the distribution of phenotypic variation of a disorder. Although not every affected individual will have the same findings, variable expressivity is often confused with reduced penetrance. The absence of a disease phenotype in an obligate carrier (ie, reduced penetrance) is not considered variable expression of the condition.
Genes that have more than one discernible effect on the phenotype are said to be pleitropic. A good example of a gene that has pleiotropic effects is the cystic fibrosis transmembrane regulator (CFTR). CFTR encodes a protein that forms cyclic-AMP–regulated chloride ion channels that span the cell membrane of specialized epithelial cells such as those that line the lungs and bowel. Mutations in CFTR result in abnormalities of the lungs, pancreas, and sweat glands. Other examples of genetic conditions that result from mutations in genes with pleiotropic effects include Marfan syndrome (characterized by ocular, cardiovascular, and skeletal defects) and osteogenesis imperfecta, in which the bones, teeth, and sclerae are affected. Thus pleiotropy can be caused by genes whose products play similar roles in different tissues and organs. Pleiotropy can also be caused by genes whose protein products play varied roles in different developmental programs. For example, individuals with campomelic dysplasia have a skeletal dysplasia that is sometimes accompanied by sex reversal. Campomelic dysplasia is caused by mutations in a gene called SRY-related HMG-BOX 9 (SOX9), which plays an important regulatory role in skeletal development and sexual differentiation.
Some genetic conditions seem to display an earlier age of onset and/or more severe expression in the more recent generations of a pedigree. This effect is called anticipation. Some investigators had proposed that anticipation was an artifact of better observation and diagnostic tools available to the contemporary clinician: a disorder previously diagnosed at age 60 might now be diagnosed at age 40. Within the past 10 years, it has been demonstrated that for some disorders, anticipation has a biological basis. 
MONOGENIC CONDITIONS: SINGLE-GENE DISORDERS
 AUTOSOMAL-DOMINANT DISORDERS
AUTOSOMAL-DOMINANT DISORDERS
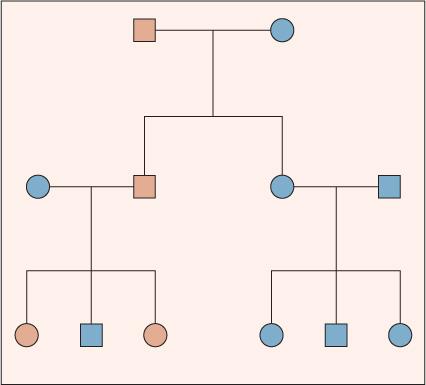
For autosomal-dominant conditions, the presence of only one copy of an altered allele at a locus is sufficient to produce a disease phenotype. Genetic disorders that are inherited in an autosomal-dominant pattern are the most common single-gene disorders described in humans. Individually, however, each autosomal-dominant disorder is relatively uncommon. Thus, most affected offspring are produced from the mating of an affected parent and an unaffected parent or from a de novo mutation. A parent affected with an autosomal-dominant disorder can transmit either the normal or altered allele to his or her offspring. Each of these events has a probability of 0.5. Thus, on average, half of the children will be heterozygous for the altered allele and express the disease, and half will be homozygous for a normal allele. Matings between individuals affected by the same autosomal-dominant disorder are rare.
An idealized pedigree for autosomal-dominant inheritance (Fig. 170-1) illustrates several important characteristics: First, the two sexes exhibit the trait in approximately equal proportions, and males and females are equally likely to transmit the trait. Second, no generation is skipped. Every individual with the trait has an affected parent. Third, father-to-son transmission is observed. Although father-to-son transmission is not required to establish autosomal-dominant inheritance, its presence excludes some other modes of inheritance (eg, X-linked and mitochondrial). Last, an affected individual transmits the trait to half of her or his offspring, on average.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


