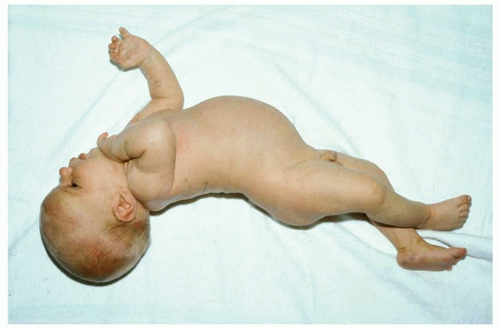
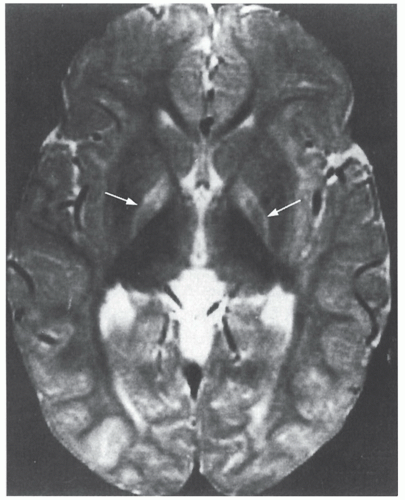
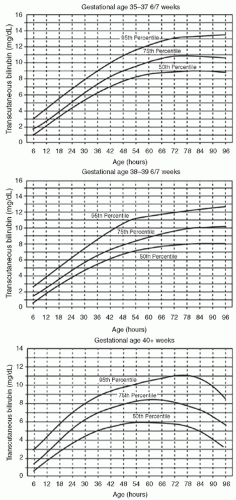
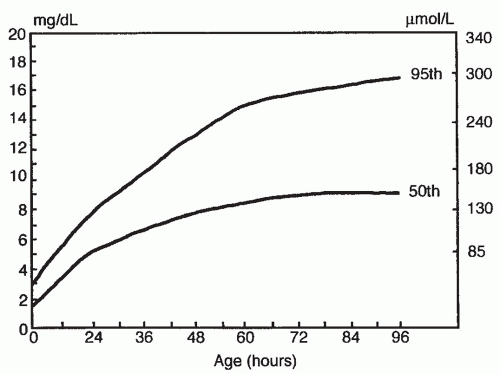
Bilirubin Levels and Developmental Outcome
Although there is no doubt about the relationship between elevated TSB levels and brain damage, the ability of a single peak bilirubin level to predict long-term neurodevelopmental outcomes is relatively poor (
117,
118,
119). In addition, our conclusions regarding hyperbilirubinemia and neurodevelopmental outcomes are limited by the quality of the published data. In many case reports of kernicterus, analyzed in detail by Ip et al. (
117,
118), it is impossible to know if the reported peak TSB levels (measured in some cases more than 7 days after birth) were true peak levels. In addition, many cohort studies have significant problems with dropouts and with blinding of the examiners (
117,
118,
120). For a detailed analysis of the published literature from 1966 to 2001 dealing with these issues, see the evidence reports of Ip et al. (
117,
118).
Hsia et al. (
121) and Mollison and Cutbush (
122) first established the link between bilirubin levels and brain damage in the early 1950s, when they demonstrated that the risk of kernicterus in infants with Rh hemolytic disease increased dramatically with rising bilirubin levels and that exchange transfusion could markedly reduce the risk. Subsequent studies suggested that, in untreated infants with hemolytic disease, the incidence of kernicterus was much higher than the incidence in markedly jaundiced infants without hemolytic disease (
123). Starting in 1967 and continuing into the late 1970s, reports from the Collaborative Perinatal Project (CPP), a study of 53,000 pregnant women and their offspring, linked moderate elevations of neonatal serum bilirubin to lower developmental scores, lower IQ scores, and an increased risk of neurologic abnormalities. These findings occurred at TSB levels previously presumed to be safe and suggested that acute bilirubin encephalopathy or classic kernicterus was only the most obvious and extreme manifestation of a spectrum of bilirubin toxicity. At the other end of the spectrum might lie more subtle forms of neurotoxicity that occur at much lower bilirubin levels and in the absence of any obvious abnormal clinical findings in the neonatal period (
105,
118,
124,
125).
Effect of Hemolysis
Newman et al. (
126) evaluated 140 term and late preterm infants with TSB levels ≥25 mg/dL. One hundred and thirty-two (94%) infants were followed for at least 2 years and 82 (59%) for 5 years with blinded neurodevelopmental evaluations. They found no difference in neurodevelopmental outcomes between the hyperbilirubinemic infants and controls. But nine children in the hyperbilirubinemia group who had positive direct antiglobulin tests (DATs) had full-scale IQ scores that were, on average, 17.8 points below those with a negative DAT. In a reanalysis of the data from the Collaborative Perinatal Project, Kuzniewicz and Newman (
127) found that the maximum TSB level was not a significant predictor of IQ scores, but there was a significant interaction term of -6.7 full-scale IQ points for those with a positive DAT and a TSB ≥25 mg/dL. In a study of full-term Turkish infants (
128), those who had DAT-positive ABO incompatibility or Rh immunization (vs. infants without hemolytic disease) had a greater risk of neurologic abnormalities and lower IQ scores when their indirect reacting bilirubin levels exceeded 20 mg/dL (342 &mgr;mol/L). Two hundred and fortynine newborns were admitted to the Cairo University Children’s Hospital with TSB levels of ≥25 mg/dL (
129), and 44 (17.7%) had evidence of moderate or severe acute bilirubin encephalopathy (ABE). The presence of Rh incompatibility (OR 48.6) and sepsis (OR 20.6) increased the risk of neurotoxicity. These data reinforce the belief that isoimmune hemolysis is an important risk factor in bilirubin-dependent brain damage although the mechanism for this association remains to be elucidated.
The hemolysis associated with G6PD deficiency and hyperbilirubinemia is also considered to be an additional risk factor for poor developmental outcome (
130), although an analysis of the cases in the U.S. Kernicterus Registry did not find this to be the case (
119).
Infants without Hemolysis
Two issues in neonatal medicine that consistently generate controversy are the relationship between hyperbilirubinemia and adverse developmental outcome in nonhemolyzing newborns and the indications for treating these infants. These issues are addressed in multiple studies, and the reader is referred to extensive reviews for details of the individual studies (
117,
118,
123,
131,
132). Although in many studies of otherwise-healthy neonates without hemolytic disease, there is no convincing demonstration of any adverse effect of TSB levels below 25 mg/dL on IQs, definite neurologic abnormalities, or sensorineural hearing loss (
117,
118,
123,
126,
131,
132,
133,
134), some older (
135), and more recent studies (
136,
137,
138,
139) have raised the possibility of subtle developmental abnormalities in some infants with modest degrees of hyperbilirubinemia.
No studies have looked specifically at infants who are 35 to 37 weeks of gestation, although the data analyzed by Ip et al. (
117,
118) include infants of 34 weeks and older. In the very large CPP, the study population included all infants with birth weights ≥2,500 g (
123,
135). Presumably, some of these infants were in the 34- to 37-weeks’ gestational age category. When they combined both abnormal and suspicious neurologic examination results, Newman and Klebanoff (
135), in their analysis of the CPP, did demonstrate a significant increase in abnormalities associated with increasing bilirubin levels. The “suspicious” abnormalities included nonspecific gait abnormalities, awkwardness, an equivocal Babinski reflex, abnormal cremasteric reflex, abnormal abdominal reflex, failure of stereognosis, questionable hypotonia, and gaze abnormalities. When the abnormal and suspiciously abnormal children were combined, the risk of abnormalities increased from 14.9% for those whose TSB levels were less than 10.0 mg/dL (171 &mgr;mol/L) to 22.4% for those whose TSB levels exceeded 20 mg/dL (340 &mgr;mol/L). Because 41,324 infants were enrolled in this study, these differences were statistically highly significant, but this finding should be kept in perspective. Even if the relationship between these findings is causal, we have no evidence that the use of a bilirubin-lowering intervention, such as phototherapy, at these low bilirubin levels would affect the outcome. Finally, as the authors point out (
135), even if bilirubin levels had been prevented from exceeding 10 mg/dL (171 &mgr;mol/L) in every infant, the expected rate of abnormal or suspicious neurologic examination results would only decline from 15.13% to 14.85%.
Less-reassuring information is provided by a study of a group of Israeli army draftees (
n = 1,948) in which their preinduction psychological and physical examinations at age 17 years were matched with their newborn bilirubin levels. Seidman et al. (
140) found an association between the risk of an IQ below 85 and a TSB level higher than 20 mg/dL (342 &mgr;mol/L) in full-term boys (but not with girls) with a negative Coombs test (
p = 0.01). No association was found between bilirubin levels and mean IQ score, the risk of physical or neurologic abnormality, or hearing loss. Three more recent studies identified “soft signs” of neurologic dysfunction in infants exposed to moderate levels of bilirubin (
137,
138,
139). Dutch investigators (
139) noted that five of eight (63%) infants at age 1 year who, as newborns, had TSB levels between 19.6 and 26 mg/dL (335 and 444 &mgr;mol/L) demonstrated minor abnormalities in muscle tone and posture compared with 0 of 20 control infants (
p < 0.001). This group subsequently evaluated 42 healthy term infants whose neonatal TSB levels were ≥12.9 mg/dL (
137), and 68 controls. A neurologic examination at age 18 months with the
examiner blinded to the history and TSB level found no difference in the incidence of minor neurologic dysfunction (MND) between the two groups, but a TSB level of ≥17.5 mg/dL was associated with an increased risk of complex MND (adjusted OR 4.21, 95% CI: 1.02 to 17.37). In a German study, children at 7 years of age whose neonatal TSB levels were greater than 20 mg/dL (342 &mgr;mol/L) scored significantly worse on a scale designed to measure choreiform and athetoid movements. Eight of sixteen (50%) children in the hyperbilirubinemia group versus 3 of 18 (17%) in the control group had abnormal scores (data not found in the original paper but kindly provided by the authors) (
120,
138).
In Milwaukee, 39/93 infants whose TSB levels as neonates were greater than 20 mg/dL were followed to ages 2.5 to 3.5 years and compared with nonjaundiced controls (
141). No differences were observed between the two groups with regard to the Bayley mental or psychomotor development indices, expressive or receptive speech, abnormal hearing, or minor neurologic abnormalities. The sample size was small, however, and in every one of these areas, the jaundiced infants performed worse. Although the differences did not achieve statistical significance, the possibility of a type II error is high.
In Northern California, Newman et al. (
126) identified 140 infants with neonatal TSB levels of ≥25 mg/dL, 10 of whom had TSB levels of ≥30 mg/dL, and 492 randomly selected controls. One hundred and thirty-six infants received phototherapy, and five had exchange transfusions. One hundred and thirty-two (94%) infants were followed for at least 2 years and 82 (59%) for 5 years with blinded neurodevelopmental evaluations by child psychologists and neurologists and detailed developmental and behavioral evaluations using parent questionnaires. There was no difference in neurodevelopmental outcomes, between the hyperbilirubinemic infants and controls although, as noted above (see
Effect of Hemolysis), hyperbilirubinemic infants with a positive DAT had lower cognitive scores but did not have more neurologic or behavioral problems.
Wong et al. (
134) studied 99 full-term Chinese neonates with nonhemolytic TSB levels of 16.8 to 29.2 mg/dL. All received phototherapy and three an exchange transfusion. These infants had regular physical, neurologic, visual, and auditory assessments every 3 to 6 months until the age of 3 years. All except two infants with mild motor delay had normal neurodevelopmental status at age 3 years. Most recently, Vandborg et al. (
133) had parents complete the Ages and Stages Questionnaire in 162 infants, ≥35 weeks of gestation and aged 1 to 5 years, whose neonatal TSB levels were ≥25 mg/dL. When compared with 146 controls, they found no evidence of developmental delay in the hyperbilirubinemic infants.
Duration of Exposure to Hyperbilirubinemia
It is intuitive to assume that the longer one is exposed to a potential toxin, the more likely it is for the toxin to have an effect. In the study from Turkey mentioned above (
128), the risk of neurologic damage increased from 2.3% to 18.7% and 26% in those infants exposed to indirect bilirubin level of ≥20 mg/dL for less than 6 hours, 6 to 11 hours, and ≥12 hours, respectively. In a study of 83 term and late preterm infants exposed to indirect bilirubin levels of ≥15 mg/dL (
142), there was a linear increase in abnormal neurologic findings from those exposed for less than 1 day (5%) to 65% in those exposed for ≥6 days. These and other data (
143) suggest that the duration of hyperbilirubinemia is related to the risk of long-term neurodevelopmental outcome although this was not seen in the NICHHD collaborative phototherapy study (
144) where no association was found between IQ and duration of exposure to bilirubin.
Bilirubin-Binding Capacity, Kernicterus, and Developmental Outcome
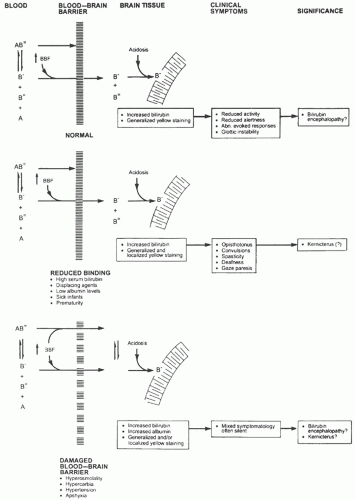
Because bilirubin that is “free” or loosely bound to albumin is more likely to cross the BBB (see
Bilirubin Chemistry and Neurotoxicity and Blood-Brain and Blood-Cerebrospinal Fluid Barriers above), our ability to predict the risk of bilirubin encephalopathy should be improved by measurement of unbound bilirubin or the reserve albumin-binding capacity (
119). Wennberg et al. (
119) have summarized the limited clinical data in term and preterm infants that suggest the measurement of free bilirubin is better than TSB as a predictor of bilirubin toxicity and, if nothing else, would reduce the risk of unnecessary intervention. Unfortunately, there are, currently, no bilirubin-binding tests in routine clinical use in the United States, although a semiautomated peroxidase method has been used in Japan (
119). In addition, there are problems with the accuracy and standardization of free bilirubin measurements and the potential for interference from photoisomers (
146).
Hearing Loss and Audiometric-Evoked Responses
The BAER test is an accurate and noninvasive means of assessing the functional status of the auditory nerve and the brainstem auditory pathway.
e-Figure 32-2 shows the BAER tracing of a normal full-term infant. The three positive waveforms labeled in the figure are those most easily identified in the neonate (
87). The latency for wave I represents the peripheral conduction time. Latency of waves III and V and the interpeak latency of waves I to III, III to V, and I to V all represent measurements of central conduction time. The interpeak latency I to V is referred to as the brainstem conduction time. Reports also include amplitudes of the waveforms. These may decrease or be lost in response to various insults (
20,
113).
Several studies have documented a relationship between TSB levels and the BAER (
87), and the acute changes seen in the BAER can be reversed by lowering the TSB level with phototherapy or exchange transfusion (
147). Abnormalities of the BAER are more closely related to unbound bilirubin levels than to TSB level (
45,
148).
As noted above (
Kernicterus, Auditory Abnormalities), deficits in central hearing, speech, and language can occur in the absence of pure-tone hearing loss, and these may be manifestations of
auditory neuropathy or
dyssynchrony (
149). This entity is defined as an abnormal or absent BAER with normal inner ear function as tested by cochlear microphonic responses or OAEs (
149). Remarkably, in a follow-up of 36 children with the Crigler-Najjar syndrome, none had evidence of sensorineural hearing loss (
150).
Hyperbilirubinemia, Autism Spectrum Disorder, and Attention Deficit Disorder
Some authors have identified an association between hyperbilirubinemia, autism, and attention deficit disorder (ADD) (
152,
153), but others have not (
154). In a population-based cohort of infants ≥35 weeks in Nova Scotia, Jangaard et al. (
136) identified 3,779 infants (6.7%) with TSB levels of ≥13.5 mg/dL (moderate hyperbilirubinemia) and 348 (0.6%) with TSB ≥19 mg/dL (severe hyperbilirubinemia). When compared with nonjaundiced controls, there was no increase in cerebral palsy, developmental delay, or deafness in infants with TSB ≥19 mg/dL, but those with moderate hyperbilirubinemia had an increased risk of developmental delay (adjusted RR: 1.6; 95% CI: 1.3 to 2.0). There was also a significant increase in the risk of attention deficit disorder in those exposed to a TSB of ≥19 mg/dL (adjusted RR: 1.9; 95% CI: 1.1 to 3.3) and a nonsignificant increase in the risk of autism (adjusted RR: 1.6; 95% CI: 1.0 to 2.5). In response to this study, Kuzniewicz et al. (
155) evaluated the data from the Northern California Kaiser Permanente Medical Care Program of infants ≥34 weeks. They found no association between TSB levels and a diagnosis of ADD. Using the same database, Croen et al. (
154) compared 338 children with the diagnoses of autism spectrum disorder (ASD) with 1,817 controls and also found no association between neonatal hyperbilirubinemia and ASD. The association between ASD and hyperbilirubinemia described by Maimburg et al. (
152) was subsequently retracted (
156). In their comprehensive review and metaanalysis, Gardener et al. (
153) evaluated the association with ASD of over 60 perinatal and neonatal factors. Some 17 risk factors including hyperbilirubinemia were associated with an increased risk of ASD. They conclude that “There is insufficient evidence to implicate any one perinatal or neonatal factor in autism etiology, although there is some evidence to suggest that exposure to a broad class of conditions reflecting general compromises to perinatal and neonatal health may increase the risk.” It is important to remember that analysis of very large databases will often identify associations that might be spurious or real, and if real, might not represent a cause-effect relation (
157).