Intersex States
Pramod P. Reddy
William R. DeFoor
Curtis A. Sheldon
Division of Pediatric Urology, Cincinnati Children’s Hospital Medical Center, University of Cincinnati, Cincinnati, Ohio 45229.
Division of Pediatric Urology, Cincinnati Children’s Hospital Medical Center, University of Cincinnati, Cincinnati, Ohio 45229.
Division of Pediatric Urology, Cincinnati Children’s Hospital Medical Center, University of Cincinnati, Cincinnati, Ohio 45229.
There are very few conditions in the practice of medicine that are as highly emotionally charged as is the topic of intersex. Almost every aspect related to intersex conditions, especially the role of the physicians and parents in gender assignment are controversial. Over the past few years, the philosophy that has guided the management of these conditions has been the center of an ongoing debate as to what is the best option from the patients’ perspective. In spite of the ongoing controversies, physicians caring for a child born with ambiguous genitalia and its parents have to ensure the child receives appropriate evaluation and that the parents are given the opportunity to make an informed and educated choice with regard to gender selection. Options should include the choice of no intervention until the child is old enough to participate in its own care.
Nowhere in the craft of surgery does an in-depth understanding of the pertinent basic sciences contribute so greatly to one’s approach to diagnosis and therapy as in the surgical management of intersex states. This complex discipline requires prompt and accurate decision making with regard to one of the most important determinants of a child’s future, his or her sex of rearing.
The social impact of giving birth to a child with ambiguous genitalia is overwhelming to the family and must be approached with reassurance and compassion and considered a social emergency (often coexisting conditions can also render this condition a medical emergency). The parents are immediately faced with the first question asked of any new parent: Is it a boy or a girl? Extensive guidance is necessary from the outset in helping the parents cope with the pressures applied by family and friends.
Once the initial dilemma has been addressed, the parents and a multidisciplinary team of physicians (that includes neonatologists, pediatric endocrinologists, obstetricians, pediatric reconstructive surgeons, geneticists, psychologists, and radiologists) must address multiple issues related to potential adverse sequelae, life-threatening metabolic complications, the risk of malignancy, the potential impact of inadequate genitalia, the psychological impact of the diagnosis on the child with maturation, and finally, the risk that further children may be born with similar malformations. After completion of the evaluation, a family care conference with the family is essential; the family is presented with all the data and the consensus recommendation of the team is offered. However, it is the parents who must ultimately decide the gender assignment for their child.
DEVELOPMENT OF SEXUAL CHARACTERISTICS
The sex of an individual may be defined by a number of criteria; however, there are three main criteria that determine the gender of an individual. These include:
Genetic sex (chromosomal sex). Determined at fertilization, based on the nature of the sperm (X or Y) fertilizing the ovum. XX is considered female genetic sex, and XY is considered male genetic sex.
Gonadal sex. The genetic sex determines the differentiation of the undifferentiated gonadal ridge into either testis (in the presence of a Y chromosome) or ovaries (the default state, in the absence of a Y chromosome)
Phenotypic sex. The developing gonads synthesize various hormones that drive the development of the external genitalia (phenotypic sex).
Of these, the most important is the phenotypic sex, for this alone determines whether an individual can experience adequate sexual intercourse and often determines if healthy, nurturing adult relationships are achievable.
The genetic considerations upon which the current understanding of mammalian sexual differentiation is based were originally described by Alfred Jost. Anomalies
of chromosomal composition may profoundly influence sexual development and are best understood with reference to the biology of cell division, which has been reviewed in detail by Simpson and Rebar (1) and Grumbach and Conte (2). The complex nature of cell division also provides potential for the generation of chromosomal anomalies. Common abnormalities in chromosomal number include aneuploidy and mosaicism. Aneuploidy refers to any deviation from the expected number of chromosomes. Proposed mechanisms include meiotic or mitotic nondisjunction and anaphase lag. Nondisjunction during meiosis results in aneuploid gametes; with fertilization, the resultant zygote is aneuploid, with an identical chromosomal constitution in all cells. Nondisjunction during mitosis produces two or more cell lines in the zygote, which is referred to as mosaicism (3). Aneuploid states associated with abnormal sexual development include 47,XXY (Klinefelter syndrome), 45,X (Turner syndrome), and 45,X/46,XY mosaicism associated with gonadal dysgenesis. The existence of two or more cell lines within an individual is termed chimerism. Presumably, 46,XX/46,XY and 46,XX/47,XXY true hermaphroditism may arise from mosaicism or chimerism. Structural chromosomal abnormalities are not uncommon and include deletions (loss of any part of a chromosome), translocations (the transfer of chromosomal material from one chromosome to another), isochromosomes (chromosomes with identical arms, arising from horizontal rather than vertical centromere division), dicentric chromosomes (a chromosome with two centromeres), ring chromosomes, and duplications. 46,XX sex-reversed males and 46,XX true hermaphrodites may represent examples of translocation of Y chromosomal material, whereas 46,XY sex-reversed females may represent examples of deletion of Y chromosomal material. An isochromosome involving the long arm of the X chromosome, 46,X,i(Xq), may be associated with gonadal dysgenesis, whereas dicentric chromosomes are occasionally seen in 45,X/46,XY mosaicism. On a molecular level, alterations in DNA sequence may result in sufficient genetic alteration to compromise gene product function and produce abnormal sexual development (4) (Table 101-1).
of chromosomal composition may profoundly influence sexual development and are best understood with reference to the biology of cell division, which has been reviewed in detail by Simpson and Rebar (1) and Grumbach and Conte (2). The complex nature of cell division also provides potential for the generation of chromosomal anomalies. Common abnormalities in chromosomal number include aneuploidy and mosaicism. Aneuploidy refers to any deviation from the expected number of chromosomes. Proposed mechanisms include meiotic or mitotic nondisjunction and anaphase lag. Nondisjunction during meiosis results in aneuploid gametes; with fertilization, the resultant zygote is aneuploid, with an identical chromosomal constitution in all cells. Nondisjunction during mitosis produces two or more cell lines in the zygote, which is referred to as mosaicism (3). Aneuploid states associated with abnormal sexual development include 47,XXY (Klinefelter syndrome), 45,X (Turner syndrome), and 45,X/46,XY mosaicism associated with gonadal dysgenesis. The existence of two or more cell lines within an individual is termed chimerism. Presumably, 46,XX/46,XY and 46,XX/47,XXY true hermaphroditism may arise from mosaicism or chimerism. Structural chromosomal abnormalities are not uncommon and include deletions (loss of any part of a chromosome), translocations (the transfer of chromosomal material from one chromosome to another), isochromosomes (chromosomes with identical arms, arising from horizontal rather than vertical centromere division), dicentric chromosomes (a chromosome with two centromeres), ring chromosomes, and duplications. 46,XX sex-reversed males and 46,XX true hermaphrodites may represent examples of translocation of Y chromosomal material, whereas 46,XY sex-reversed females may represent examples of deletion of Y chromosomal material. An isochromosome involving the long arm of the X chromosome, 46,X,i(Xq), may be associated with gonadal dysgenesis, whereas dicentric chromosomes are occasionally seen in 45,X/46,XY mosaicism. On a molecular level, alterations in DNA sequence may result in sufficient genetic alteration to compromise gene product function and produce abnormal sexual development (4) (Table 101-1).
Every individual begins with common, indifferent gonadal and genital primordia. An inherent tendency to feminize dominates unless specific developmental influences are present to direct male morphogenesis. Three specific but closely interrelated developmental sequences are required for normal sexual development, each governed by unique embryologic influences. These are gonadal, genital ductal, and external genitalia development. By weeks 5 to 7, the chromosomal sex determines the gonadal sex through a cascade of molecular events that have been extensively studied since the mid-1950s and understood since the mid-1990s. Because the presence of the Y chromosome was associated with the development of testis, it was extensively studied to identify the testis determining factor (TDF). A variety of candidates were believed to be the definitive TDF; these included the H-Y antigen and Zinc-finger Y gene (ZFY). The discovery of the sex determining region of the Y chromosome (SRY gene) in humans and mice in 1990 has provided the most likely candidate to date for the TDF (5,6). The role of the SRY gene was confirmed by experiments performed by Koopman et al. in 1991, in which a 14-kb DNA fragment from the SRY gene was inserted into the genome of female mouse embryos; these mice then developed into males (7,8).
TABLE 101-1 Chromosomal Location of Genes Pertinent to Sexual Development. | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The SRY gene is located on the Y chromosome, between the ZFY gene and the pseudoautosomal region, comprising of a single exon containing 237 bp. The SRY gene functions as a transcription factor and is believed to stimulate other genes that are required for testicular development and the determination of male gonadal sex by unraveling the DNA double helix. There are other genes that play a role in determining the gonadal differentiation at the various stages. These include genes that act on the intermediate mesoderm and cause it to differentiate into the indifferent gonad [i.e., WT-1 gene (9), DAX-1 gene (Dss-Ahc critical region of X chromosome) (10)] and genes that have two sites of action (both at the level of the intermediate mesoderm and the indifferent gonad) steroidogenic factor-1 (SF-1) (11,12), and SOX-9 gene (13,14). These genes in addition to the SRY gene are all expressed at various stages of development of the urogenital ridge.
Strong evidence exists to suggest that other genes are required to affect testicular organogenesis, with both X chromosomal and autosomal influences implicated. These could be involved in SRY regulation or could be downstream targets of SRY. Data exist to suggest that some 46,XX males and 46,XX true hermaphrodites may in fact be negative for SRY (15,16,17,18). In addition, individuals with
46,XY gonadal dysgenesis are reported with present and unaltered SRY sequences (19,20,21,22). Such determinations are difficult to interpret; however, given the observations that potentially undetected mosaicism with a Y-bearing cell line may explain some cases of 46,XX males (15) and that postzygotic SRY mutations in portions of gonadal tissue may result in 46,XY true hermaphroditism (23). Vilain and colleagues (24) analyzed the histologic findings in a series of patients with 46,XY gonadal dysgenesis. Individuals with streak gonads composed of exclusively ovarian-like stroma were found to have SRY mutations, whereas those with streak gonads containing undifferentiated stroma harboring either tubules or a rete structure had no detectable mutation in the SRY gene.
46,XY gonadal dysgenesis are reported with present and unaltered SRY sequences (19,20,21,22). Such determinations are difficult to interpret; however, given the observations that potentially undetected mosaicism with a Y-bearing cell line may explain some cases of 46,XX males (15) and that postzygotic SRY mutations in portions of gonadal tissue may result in 46,XY true hermaphroditism (23). Vilain and colleagues (24) analyzed the histologic findings in a series of patients with 46,XY gonadal dysgenesis. Individuals with streak gonads composed of exclusively ovarian-like stroma were found to have SRY mutations, whereas those with streak gonads containing undifferentiated stroma harboring either tubules or a rete structure had no detectable mutation in the SRY gene.
In the absence of a Y chromosome, ovarian differentiation generally occurs. However, the X chromosomes play a critical role in ovarian organogenesis. Individuals with monosomy X (45,X) demonstrate early ovarian development. In the absence of a second X chromosome, oocytes usually do not survive meiosis, and folliculogenesis fails to occur or is defective. The result is dysgenetic (streak) gonads. Because similar events are noted for individuals with deletion of either the short or long arm of the X chromosome, both arms are believed to contain important ovarian maintenance determinants. As reviewed by Simpson and Rebar (1), individuals with deletions in the Xp11.2–11.4 and Xq11.3–21 regions create a high risk for primary amenorrhea. Deletions in the Xp21 and Xq25–26 regions also cause significant ovarian dysfunction. In each instance, the more proximal region is where the most critical genes are located.
A unique event occurs involving the X chromosome, the formation of the Barr body. In 1949, Barr and Bertram described a chromatin mass located at the periphery of the nucleus and found in female but not in male cells (25,26). In the past, this observation was used clinically to help suggest an infant’s chromosomal content and aid in the evaluation of intersex states. An analysis of buccal mucosal smears was employed. A female 46,XX karyotype was generally associated with at least a 20% identification rate of Barr bodies within the nuclei (chromatin-positive). Barr bodies are also identifiable in 47,XXY males, and multiple Barr bodies may be identified in both males and females with more than two X chromosomes. Conversely, a relative absence of Barr bodies implied a 46,XY karyotype, but could also be seen in individuals with 45,X and 45,X/46,XY. Such individuals are referred to as chromatin-negative.
Gonadal Development
Early in embryogenesis, the gonads of both sexes are both indifferent and bipotential. Germ cells are identifiable in the dorsal endoderm of the yolk sac near the origin of the allantois. During weeks 4 and 5 of gestation, they migrate along the dorsal mesentery of the hindgut to the gonadal ridges. Invasion of the underlying mesoderm during week 6 allows them to become incorporated into the developing primary sex cords. These events are depicted in Fig. 101-1. At approximately 40 days of gestation, divergent gonadal differentiation becomes evident (27) (Fig. 101-2).
In the human testis, seminiferous cords are differentiated by 7 weeks. Differentiation of the supporting cells is observed during weeks 8 to 11. Sertoli cells demonstrate ultrastructural evidence of intense protein biosynthesis as early as 8 weeks. The product of this biosynthesis would appear to be the müllerian inhibiting substance (MIS) as evidenced by the observation of coincident regression of müllerian ducts and the detection of MIS in the rough endoplasmic reticulum (28). The multipotent Sertoli cell is also believed to secrete inhibin [provides negative feedback for follicle-stimulating hormone (FSH) secretion], activin (provides positive feedback for FSH secretion), and follistatin (provides inhibition of FSH release), and in addition is believed to nurture the germ cells and is postulated to prevent meiosis, possibly by the production of a meiosis-inhibiting substance.
Similarly, Leydig cells acquire ultrastructural features that suggest active steroid synthesis as early as 8 weeks, coincident with the onset of testosterone formation and metabolism in the human fetus (29,30). Human chorionic gonadotropin (hCG)-luteinizing hormone (LH) cell membrane receptors are detected by week 12 of gestation (31,32). Peak testosterone levels in the human fetus circulation is reached at approximately 16 weeks (31,33). It is suggested that hCG secreted by the syncytiotrophoblast stimulates testosterone secretion during this critical period of male sexual development (34). The dependence of fetal testosterone biosynthesis on placental hCG has, however, been brought into question (32,35).
In the female fetus, ovigerous cords are noted during week 7 (27). Oogonia are surrounded by cuboidal cells with long overlapping cytoplasmic processes that are believed to be precursors of granulosa cells. This early cytologic differentiation is matched by the ability of the human fetal ovary to aromatize androgens, documented as early as 8 weeks of gestational age (36). Similar to the Sertoli cell, the granulosa cell is found to produce inhibin, activin, and follistatin. In contrast, however, the fetal ovary does not contain hCG receptors nor detectable quantities of MIS.
Development of the Internal Genital Ductal Systems
The development of the genital ductal systems and urinary tract has several critical anatomic and functional correlates. Figure 101-3 diagrammatically depicts these events. From the intermediate mesoderm arises the nephrogenic cord from which the mesonephros (the temporary kidney) and metanephros (the permanent kidney) develop. Cranially, the nephrogenic cord gives rise to the mesonephros
as mesonephric vesicles develop that are connected by mesonephric tubules to the mesonephric duct. The latter develops initially as a solid rod beneath the surface ectoderm, ultimately opening into the developing cloaca. Within the mesonephros, tufts of capillaries give origin to primitive glomeruli, which begin to produce urine by week 6 of gestation.
as mesonephric vesicles develop that are connected by mesonephric tubules to the mesonephric duct. The latter develops initially as a solid rod beneath the surface ectoderm, ultimately opening into the developing cloaca. Within the mesonephros, tufts of capillaries give origin to primitive glomeruli, which begin to produce urine by week 6 of gestation.
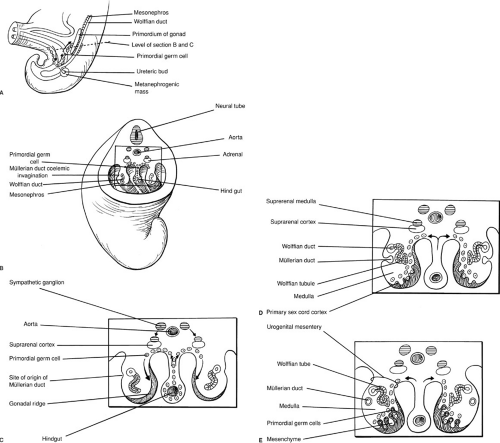 FIGURE 101-1. (A) Migration of primordial germ cells in 5-week embryo. (B, C) Transverse views of germ cell migration. (D) Six-week embryo with developing müllerian ducts and primary sex cords. (E) Further development of the müllerian and wolffian ducts as well as the primary sex cords. Arrows indicate primordial germ cell migration. (From Moore KL. The developing human: clinically oriented embryology, 3rd ed. Philadelphia: WB Saunders, 1982:271, with permission.) |
Mesonephric differentiation occurs in a cranial-to-caudal direction, during which the more cranially located nephrons progressively degenerate. Further caudally, the metanephros originates from the metanephric blastema of the nephrogenic cord and the ureteric bud and its branches. The latter develops as a diverticulum from the caudal mesonephric duct and is believed to induce development of the metanephric tubules. It becomes branched, giving origin to the ureter, renal pelvis, calyces, and collecting tubules. Late in the first trimester, the mesonephros ceases to be functional and excretion is taken over by the metanephros. Only a few caudalmost mesonephric tubules persist in the male to become the efferent ductules of the testis. These ductules drain into the mesonephric (wolffian) duct, which gives origin to the epididymis, vas deferens, seminal vesicles, and ejaculatory ducts.
The paramesonephric (müllerian) ducts arise as coelomic invaginations in the mesonephros (Fig. 101-1). This process is believed to be induced by the mesonephric duct and begins cranially at a site that represents the future abdominal ostium of the uterine (fallopian) tube. The paramesonephric ducts grow caudally, fuse, and ultimately drain into the urogenital sinus. The unfused portions give rise to the fallopian tubes, whereas the fused portion develops into the uterus and upper vagina. Because müllerian duct differentiation fails to occur in the absence of a wolffian duct, renal aplasia is often associated with anomalies of the fallopian tubes, uterus, and vagina.
The differentiation of the internal genital ductal system is critically dependent on gonadal influences. Both the mesonephric and paramesonephric ducts are present in the fetus at 7 weeks’ gestation in both males and females. In the absence of testicular influence, regardless of whether an ovary is present, mesonephric ducts regress and paramesonephric ducts mature. In the presence of a testis, the paramesonephric ducts regress and the mesonephric ducts mature into the epididymis, vas deferens, and seminal vesicles. During the third month of fetal life, MIS produced by fetal Sertoli cells and testosterone
produced by fetal Leydig cells drive these changes locally, and hence, principally unilaterally.
produced by fetal Leydig cells drive these changes locally, and hence, principally unilaterally.
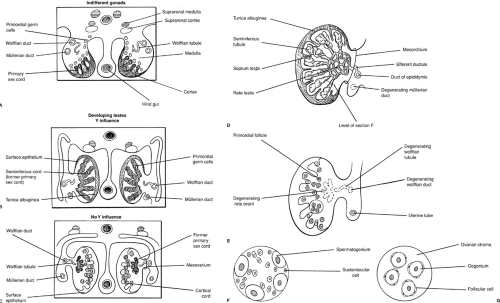 FIGURE 101-2. Ovarian and testicular differentiation. (A) Indifferent 6-week gonad. (B) Primary sex cords develop into seminiferous cords and the tunica albuginea becomes detectable at 7 weeks. (C) Twelve-week ovary. Cortical cords have extended from the epithelium. Primary sex cords are displaced into the mesovarium, forming rudimentary rete ovarii. (D) Twenty-week testis. The seminiferous tubules and rete testis have developed from seminiferous cords. Wolffian tubules develop into efferent ductules and the wolffian duct gives rise to the duct of the epididymis. (F, G) Section of the testis and ovary in the 20-week fetus. (From Moore KL. The developing human: clinically oriented embryology, 3rd ed. Philadelphia: WB Saunders, 1982:271, with permission.) |
Experimentally, gonadectomy of either male or female fetuses early in gestation results in wolffian regression and müllerian development. Further, unilateral castration of a male fetus results in ipsilateral wolffian regression and müllerian development, whereas the contralateral genital ducts develop in a normal masculine fashion (37). Conversely, testicular grafts placed in female fetuses result in müllerian regression and wolffian stimulation. The local implantation of androgens in female fetuses causes unilateral wolffian ductal maturation, whereas müllerian ductal development is unaltered (37,38).
MIS is a glycoprotein composed of two identical 72-kd subunits, connected by disulfide bonds (39), that are encoded by a gene on the short arm of chromosome 19
(40). MIS is a member of the transforming growth factor β (TGF- β) superfamily. Work by Donahoe and associates (41) suggested that MIS acts by blocking tyrosine phosphorylation on membrane proteins. MIS appears to have multiple potential functions, including not only müllerian duct regression but also germ cell maturation and gonadal morphogenesis, lung maturation, testicular descent, and growth inhibition of certain transformed cells (42,43,44). MIS is also involved in a negative feedback loop involving the Leydig cells, thereby regulating testosterone production (45,46). Hutson et al. suggested that MIS plays a role in testicular descent because it is expressed at high levels throughout childhood, with levels decreasing only after puberty (47,48).
(40). MIS is a member of the transforming growth factor β (TGF- β) superfamily. Work by Donahoe and associates (41) suggested that MIS acts by blocking tyrosine phosphorylation on membrane proteins. MIS appears to have multiple potential functions, including not only müllerian duct regression but also germ cell maturation and gonadal morphogenesis, lung maturation, testicular descent, and growth inhibition of certain transformed cells (42,43,44). MIS is also involved in a negative feedback loop involving the Leydig cells, thereby regulating testosterone production (45,46). Hutson et al. suggested that MIS plays a role in testicular descent because it is expressed at high levels throughout childhood, with levels decreasing only after puberty (47,48).
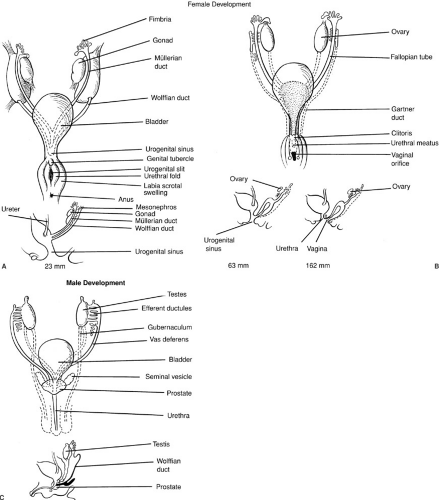 FIGURE 101-3. (A–C) Embryonic differentiation of female genital ducts (see text). (From Grumbach MM, Conte FA. Disorder of sex differentiation. In: Wilson JD, Foster DW, eds. Williams textbook of endocrinology. Philadelphia: WB Saunders, 1992:871, with permission.) |
Similarly, local androgen production by the fetal testis promotes ipsilateral wolffian duct maturation. This effect appears to require high local testosterone concentration because wolffian differentiation is not induced by systemic androgen administration in early gestation. Because 5α-reductase is not present in wolffian ducts, it is testosterone, not dihydrotestosterone, that binds to the cytosolic androgen receptor to induce these phenotypic changess (29).
These embryologic phenomena explain many observations made clinically:
Female fetuses with congenital virilizing adrenal hyperplasia do not demonstrate müllerian regression or wolffian maturation.
46,XY women with testicular feminization (complete androgen insensitivity) have regression of müllerian ducts, but wolffian ducts remain rudimentary.
Genetic males with severe defects in steroid biosynthesis that result in androgen deficiency have rudimentary wolffian derivatives.
Genetic males with isolated deficiency of MIS have persistence of müllerian derivatives, but wolffian development proceeds normally.
In individuals with asymmetric gonadal development such as 45,X/46,XY, mixed gonadal dysgenesis, and true hermaphroditism, genital ductal development follows that of the homolateral gonad; the presence of testicular tissue results in ipsilateral müllerian regression and wolffian development.
Development of the Urogenital Sinus and External Genitalia
At week 8 of gestation, the external genitalia of both males and females are identical and remain uncommitted. Figure 101-4 outlines the differentiation of male and female external genitalia. In males, the genital tubercle becomes the penis, whereas the urethral folds fuse to become the floor of the penile urethra and the corpus spongiosum that encloses the penile urethra. In analogous fashion, the labioscrotal swellings fuse in the midline to become the scrotum. In the female, the genital tubercle becomes the clitoris and the urethral folds and labioscrotal swellings do not fuse, becoming the labia minora and the labia majora, respectively.
Sexual differentiation of the urogenital sinus is illustrated in Fig. 101-5. As with the external genitalia, the urogenital sinus is similar in both sexes up to week 8 or 9 of fetal life. The müllerian tubercle protrudes from the posterior wall of the urogenital sinus between the two wolffian duct orifices. This contact of the urogenital sinus with the fused müllerian ducts is critical to normal development of the vagina (49,50). Although this interpretation is controversial, the lower vagina appears to be of urogenital sinus origin and the upper vagina of müllerian origin. Expansion of the tissue composing the vesicovaginal septum allows for spatial separation of the vagina and urethra. In the male, the urogenital sinus undergoes elongation to form the prostatic and perineal urethra.
In analogous fashion to the differentiation of the internal genital ducts, masculinization of the urogenital sinus and external genitalia requires the presence of a testis. In contrast, however, this effect occurs in response to exposure to dihydrotestosterone (DHT) and can be induced by systemic androgen exposure (Fig. 101-6). Testosterone enters the cell by diffusion, whereupon it may bind to a high-affinity androgen receptor protein, be aromatized to estradiol, or be converted to DHT by the action of 5α-reductase. Although both testosterone and DHT may bind to the androgen receptor protein, the binding affinity of the receptor for DHT is much greater. As with several members of the steroid receptor superfamily, the DNA-binding site is occupied by an inhibitory (e.g., heat shock) protein (51). Binding of the androgen to the receptor leads to dissociation of the inhibitory protein by conformational change, enabling the receptor to bind to specific sites on the DNA, referred to as receptor-dependent transcriptional enhancers. The result is transcription and processing of mRNA, which is then translated, allowing the synthesis of new proteins that mediate the androgenic effects involved with masculinization of the urogenital sinus and external genitalia.
Psychosexual Development
It has been generally assumed that gender identity (the identification of one’s self as either male or female) is largely a learned process, and that this gender identity is firmly established by 1.5 and 2.5 years of age (52). Clearly, the assignment of the sex of rearing is the single most critical event in this process. Reinforcement of this assignment is crucial and requires unambiguous genital anatomy and unambiguous family interactions. One must not, however, overlook the importance of endocrine effects during prenatal development and puberty. Gender-related behavior, including gender role, has been demonstrated to be influenced by prenatal sex hormone exposure (53,54).
Further, pubertal hormonal influences producing phenotypic changes counter to that of the original gender assignment have been reported to induce not only doubts about gender identity but to cause alterations in gender behavior in some individuals (55). Males with a small penis, followed into adulthood, do not in general experience altered gender identity, altered desire for heterosexual activity, or alterations in other activities generally considered to be predominantly male (56). This and other studies, however, have demonstrated that life with inadequate or compromised genitalia is often confounded by debilitating emotional disturbances.
Further, pubertal hormonal influences producing phenotypic changes counter to that of the original gender assignment have been reported to induce not only doubts about gender identity but to cause alterations in gender behavior in some individuals (55). Males with a small penis, followed into adulthood, do not in general experience altered gender identity, altered desire for heterosexual activity, or alterations in other activities generally considered to be predominantly male (56). This and other studies, however, have demonstrated that life with inadequate or compromised genitalia is often confounded by debilitating emotional disturbances.
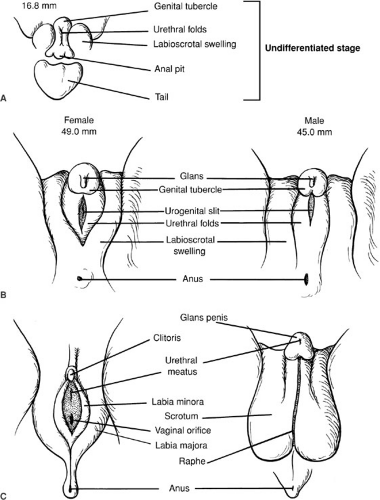 FIGURE 101-4. Sexual differentiation of the external genitalia. (A) Undifferentiated stage of development. (B) Between 8 and 12 weeks of gestation, human genital differentiation occurs. (C) Adult genitalia. (From Sizonenko PC. Sexual differentiation. In: Bertrand J, Rappaport R, Sizonenko PC, eds. Pediatric endocrinology. Baltimore: Williams & Wilkins, 1993:88, with permission.) |
Developmental Temporal Sequence
An understanding of the temporal sequence of the development of the reproductive tract is helpful clinically. As illustrated in Fig. 101-7, one may conceptualize this process as a series of interrelated steps. Chromosomal sex is determined at the time of conception. This is followed by an indifferent phase of embryogenesis that lasts approximately 7 weeks. Thereafter, divergent development is progressive with the sequential differentiation of the gonads, followed by the internal genital ducts, followed by the urogenital sinus and external genitalia.
Evidence exists to suggest that during the period of organogenesis, inclusive of the differentiation of the external genitalia, fetal Leydig cell activity is driven by placental hCG rather than by LH from the fetal pituitary (31,33). The pattern of testosterone secretion during early gestation reflects that of hCG and the fetal testicular hCG-binding capacity (31,57,58). In addition, the expression of several steroidogenic genes appears to be directly regulated by circulating hCG (30,59,60,61). Fetal pituitary LH
appears to take over the modulation of testosterone synthesis after midgestation and, along with placental hCG, appears to play a critical role in the growth of the differentiated penis and scrotum as well as descent of the testes (57).
appears to take over the modulation of testosterone synthesis after midgestation and, along with placental hCG, appears to play a critical role in the growth of the differentiated penis and scrotum as well as descent of the testes (57).
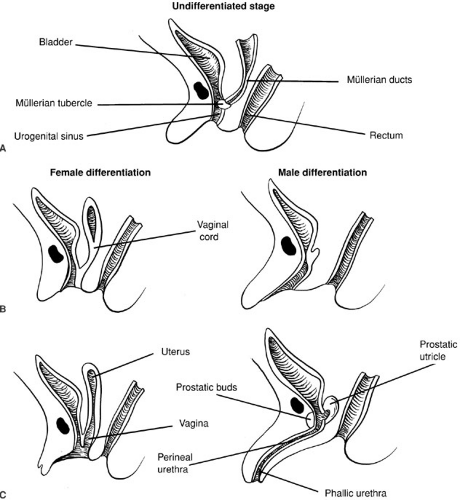 FIGURE 101-5. Sexual differentiation of the urogenital sinus. (From Sizonenko PC. Sexual differentiation. In: Bertrand J, Rappaport R, Sizonenko PC, eds. Pediatric endocrinology. Baltimore: Williams & Wilkins, 1993:88, with permission.) |
These observations, coupled with the facts that after about 12 weeks of gestation the vagina has separated from the urogenital sinus, and the urethral folds and labioscrotal swellings are unable to fuse even with intense androgenic stimulation, explain several important clinical observations: (1) females exposed to androgens after the period of organogenesis develop clitoromegaly, but urethral fold and labioscrotal fusion does not occur; (2) the diagnosis of congenital virilizing adrenal hyperplasia generally does not explain clitoromegaly in the absence of midline fusion; and (3) males with congenital hypopituitarism or selective gonadotropin deficiency often present with microphallus, but the penis and scrotum are generally well differentiated.
Summary of Reproductive Tract Differentiation
Figure 101-8 summarizes the events leading to normal sexual development in both males and females. In the presence of two normal X chromosomes, two normal ovaries develop. In the absence of testosterone and MIS, paramesonephric ducts mature into fallopian tubes, uterus, and upper vagina, whereas mesonephric ductal elements regress. Similarly, in the absence of androgenic stimulation, feminine differentiation of the urogenital
sinus and the external genitalia occurs: (1) In the absence of an X chromosome or a critical portion of an X chromosome, important ovarian developmental failure may result. In addition, 46,XX males with SRY translocations and 46,XX males with undetected mosaicism with a Y-bearing cell line are reported and are associated with testicular organogenesis. 46,XX and 46,XX/46,XY true hermaphroditism is similarly associated with development of some testicular tissue. (2) Once present, testicular tissue may produce sufficient testosterone and MIS to result in mesonephric development and paramesonephric regression, which if unilateral, is restricted to the side where testicular tissue is located. (3) Exposure of the fetus to either exogenous (e.g., maternal ingestion) or endogenous (e.g., congenital virilizing adrenal hyperplasia) androgen may result in various degrees of virilization of the urogenital sinus and the external genitalia. In the presence of a normal Y and X chromosome, testicular development follows. The production of MIS by Sertoli cells and the production of testosterone by Leydig cells results in paramesonephric regression and mesonephric development, respectively. During the period of organogenesis, Leydig cell steroid synthesis is driven by placental hCG and thereafter by fetal pituitary LH. Testosterone is converted to DHT by 5α-reductase, which, in the presence of the cytosolic androgen receptor protein encoded by a gene on the X chromosome, causes masculinization of the urogenital sinus and external genitalia. (4) In the absence of a Y chromosome (e.g., 45,X/46,XY) or (5) a critical portion of the Y chromosome (SRY), defective testicular development may occur. (6) MIS deficiency may occur in isolation or as part of more complete testicular failure and results in persistence of paramesonephric structures. (7) Leydig cell steroid synthetic failure may occur as a result of specific enzymatic deficiency or as part of more complete testicular failure and results in incomplete masculinization of the internal genital ducts, urogenital sinus, and external genitalia. (8) Partial androgen receptor deficiency may result in incomplete masculinization, whereas complete deficiency may result in a nearly normal female phenotype. Because MIS production is normal, paramesonephric structures are rudimentary. (9) 5α-Reductase deficiency results in insufficient DHT production, which produces only partial virilization. (10) Congenital hypopituitarism or isolated gonadotropin deficiency may result in inadequate stimulation of Leydig cell function. The result is a small but completely formed penis and scrotum.
sinus and the external genitalia occurs: (1) In the absence of an X chromosome or a critical portion of an X chromosome, important ovarian developmental failure may result. In addition, 46,XX males with SRY translocations and 46,XX males with undetected mosaicism with a Y-bearing cell line are reported and are associated with testicular organogenesis. 46,XX and 46,XX/46,XY true hermaphroditism is similarly associated with development of some testicular tissue. (2) Once present, testicular tissue may produce sufficient testosterone and MIS to result in mesonephric development and paramesonephric regression, which if unilateral, is restricted to the side where testicular tissue is located. (3) Exposure of the fetus to either exogenous (e.g., maternal ingestion) or endogenous (e.g., congenital virilizing adrenal hyperplasia) androgen may result in various degrees of virilization of the urogenital sinus and the external genitalia. In the presence of a normal Y and X chromosome, testicular development follows. The production of MIS by Sertoli cells and the production of testosterone by Leydig cells results in paramesonephric regression and mesonephric development, respectively. During the period of organogenesis, Leydig cell steroid synthesis is driven by placental hCG and thereafter by fetal pituitary LH. Testosterone is converted to DHT by 5α-reductase, which, in the presence of the cytosolic androgen receptor protein encoded by a gene on the X chromosome, causes masculinization of the urogenital sinus and external genitalia. (4) In the absence of a Y chromosome (e.g., 45,X/46,XY) or (5) a critical portion of the Y chromosome (SRY), defective testicular development may occur. (6) MIS deficiency may occur in isolation or as part of more complete testicular failure and results in persistence of paramesonephric structures. (7) Leydig cell steroid synthetic failure may occur as a result of specific enzymatic deficiency or as part of more complete testicular failure and results in incomplete masculinization of the internal genital ducts, urogenital sinus, and external genitalia. (8) Partial androgen receptor deficiency may result in incomplete masculinization, whereas complete deficiency may result in a nearly normal female phenotype. Because MIS production is normal, paramesonephric structures are rudimentary. (9) 5α-Reductase deficiency results in insufficient DHT production, which produces only partial virilization. (10) Congenital hypopituitarism or isolated gonadotropin deficiency may result in inadequate stimulation of Leydig cell function. The result is a small but completely formed penis and scrotum.
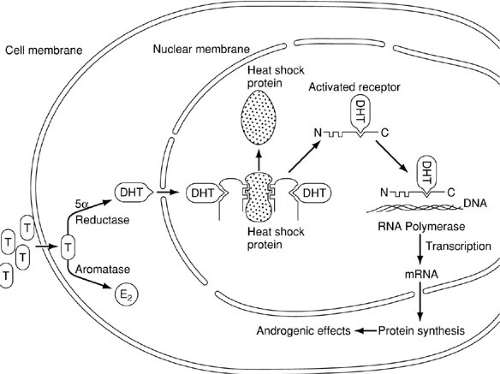 FIGURE 101-6. Mechanism of action of androgens on cell function (see text). T, testosterone; DHT, dihydrotestosterone. (From Grumbach MM, Conte FA. Disorder of sex differentiation. In: Wilson JD, Foster DW, eds. Williams textbook of endocrinology. Philadelphia: WB Saunders, 1992:875, with permission.) |
Defects in Steroid Biosynthesis
The subject of steroid biosynthesis has been extensively reviewed (2). Defects can result in either masculinization of genetic females, female sexual infantilism, undermasculinization of the genetic male, or excessive masculinization of the genetic male. These defects are often overlooked because of the wide variability of phenotypic expression. They must be understood by surgeons because the first level of referral may be to the surgeon for correction of what may appear as a relatively common anomaly such as hypospadias.
The steroid biosynthetic pathways are diagrammed in Fig. 101-9. Congenital adrenal hyperplasia (CAH) is
responsible for the majority of cases of female pseudohermaphroditism and accounts for approximately one-half of ambiguous genitalia cases. The most common enzymatic defect is 21-hydroxylase deficiency. This condition is caused by mutations in the CYP21 gene on chromosome 6p21.3. Consequently, this deficiency is inherited in an autosomal recessive fashion, and its inheritance is intimately associated with that of the major transplant antigens. Analysis of human histocompatability antigens may therefore be helpful in genetic counseling.
responsible for the majority of cases of female pseudohermaphroditism and accounts for approximately one-half of ambiguous genitalia cases. The most common enzymatic defect is 21-hydroxylase deficiency. This condition is caused by mutations in the CYP21 gene on chromosome 6p21.3. Consequently, this deficiency is inherited in an autosomal recessive fashion, and its inheritance is intimately associated with that of the major transplant antigens. Analysis of human histocompatability antigens may therefore be helpful in genetic counseling.
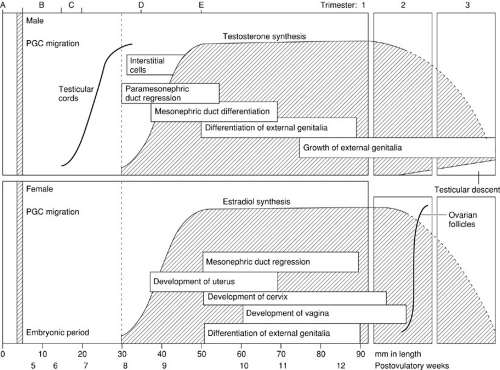 FIGURE 101-7. Prenatal development of the reproductive system as a function of embryonic/fetal length and age. (A) Chromosomal sex established. (B) Indifferent phase. (C) Gonadal differentiation. (D) Genital ductal differentiation. (E) External genitalia differentiation. (See text.) PGC, primordial germ cell. (From O’Rahilly R, Muller F. Human embryology and teratology. New York: Wiley-Liss, 1992:207, with permission.) |
Based on data from newborn screening programs for CAH, the incidence of classic 21-hydroxylase deficiency is about 1 in 14,000 newborns worldwide, with an increased incidence seen in Alaska (1 in 800), Brazil (1 in 7,500), and the Philippines (1 in 7,000). Salt-wasting CAH accounts for 66% of reported cases and simple-virilizing CAH accounts for 32% (62).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


