Hormone Biosynthesis, Metabolism, and Mechanism of Action
 |
The classical definition of a hormone is a substance that is produced in a special tissue, where it is released into the bloodstream, and travels to distant responsive cells in which the hormone exerts its characteristic effects. What was once thought of as a simple voyage is now appreciated as an odyssey that becomes more complex as new facets of the journey are unraveled in research laboratories throughout the world. Indeed, the notion that hormones are products only of special tissues has been challenged.
Complex hormones and hormone receptors have been discovered in primitive, unicellular organisms, suggesting that endocrine glands are a late development of evolution. The widespread capability of cells to make hormones explains the puzzling discoveries of hormones in strange places, such as gastrointestinal hormones in the brain, reproductive hormones in intestinal secretions, and the ability of cancers to unexpectedly make hormones. Hormones and neurotransmitters were and are a means of communication. Only when animals evolved into complex organisms did special glands develop to produce hormones that could be used in a more sophisticated fashion. Furthermore, hormones must have appeared even before plants and animals diverged because there are many plant substances similar to hormones and hormone receptors. Therefore, it is not surprising that, because every cell contains the genes necessary for hormonal expression, cancer cells, because of their dedifferentiation, can uncover gene expression and, in inappropriate locations and at inappropriate times, make hormones.
Hormones, therefore, are substances that provide a means of communication and should now be viewed broadly as chemical regulatory and signaling agents. The classic endocrine hormones travel through the bloodstream to distant sites, but cellular communication is also necessary at local sites. Paracrine, autocrine, and intracrine depict a more immediate form of communication.
Paracrine Communication
Intercellular communication involving the local diffusion of regulating substances from a cell to nearby (contiguous) cells.
Autocrine Communication
Intracellular communication whereby a single cell produces regulating substances that in turn act upon receptors on or within the same cell.
Intracrine Communication
This form of intracellular communication occurs when unsecreted substances bind to intracellular receptors; in other words, a regulating factor acts within the cell that secretes it.
Let us follow an estradiol molecule throughout its career and in so doing gain an overview of how hormones are formed, how hormones work, and how hormones are metabolized. Estradiol begins its lifespan with its synthesis in a cell specially suited for this task. For this biosynthesis to take place, the proper enzyme capability must be present along with the proper precursors. In the adult human female the principal sources of estradiol are the granulosa cells of the developing follicle and the corpus luteum. These cells possess the ability to turn on steroidogenesis in response to specific stimuli. The stimulating agents are the gonadotropins, follicle-stimulating hormone (FSH) and luteinizing hormone (LH). The initial step in the process that will give rise to estradiol is the transmission of the message from the stimulating agents to the steroid-producing mechanisms within the cells.
Messages that stimulate steroidogenesis must be transmitted through the cell membrane. This is necessary because gonadotropins, being large glycopeptides, do not ordinarily enter cells but must communicate with the cell by joining with specific receptors on the cell membrane. In so doing they activate a sequence of communication. A considerable amount of investigation has been devoted to determining the methods by which this communication takes place. E. W. Sutherland, Jr., received the Nobel Prize in 1971 for proposing the concept of a second messenger.
Gonadotropin, the first messenger, activates an enzyme in the cell membrane called adenylate cyclase. This enzyme transmits the message by catalyzing the production of a second messenger within the cell, cyclic adenosine 3′,5′-monophosphate (cyclic AMP). The message passes from gonadotropin to cyclic AMP, much like a baton in a relay race.
Cyclic AMP, the second messenger, initiates the process of steroidogenesis, leading to the synthesis and secretion of the hormone estradiol. This notion of message transmission has grown more and more complex with the appreciation of physiologic concepts, such as the heterogeneity of peptide hormones, the up- and down-regulation of cell membrane receptors, the regulation of adenylate cyclase activity, and the important roles for autocrine and paracrine regulating factors.
Secretion of estradiol into the bloodstream directly follows its synthesis. Once in the bloodstream, estradiol exists in two forms, bound and free. A majority of the hormone is bound to protein carriers, albumin and sex steroid hormone-binding globulin. The biologic activity of a hormone is limited by binding in the blood, thereby avoiding extreme or sudden reactions. In addition, binding prevents unduly rapid metabolism, allowing the hormone to exist for the length of time necessary to ensure a biologic effect. This reservoir-like mechanism avoids peaks and valleys in hormone levels and allows a more steady state of hormone action.
The biologic and metabolic effects of a hormone are determined by a cells ability to receive and retain the hormone. The estradiol that is not bound to a protein, but floats freely in the bloodstream, readily enters cells by rapid diffusion. For estradiol to produce its effect, however, it must be grasped by a receptor within the cell. The job of the receptor is to aid in the transmission of the hormone’s message to nuclear gene transcription. The result is production of messenger RNA leading to protein synthesis and a cellular response characteristic of the hormone.
Once estradiol has accomplished its mission, it is eventually released back into the bloodstream. It is possible that estradiol can perform its duty several times before being cleared from the circulation by metabolism. On the other hand, many molecules are metabolized without ever having the chance to produce an effect. Unlike estradiol, other hormones, such as testosterone, can either work directly or are metabolized and altered within the cell in which an effect is produced. In the latter case, a metabolite is released into the bloodstream as an inactive compound. Clearance of steroids from the blood varies according to the structure of the molecules.
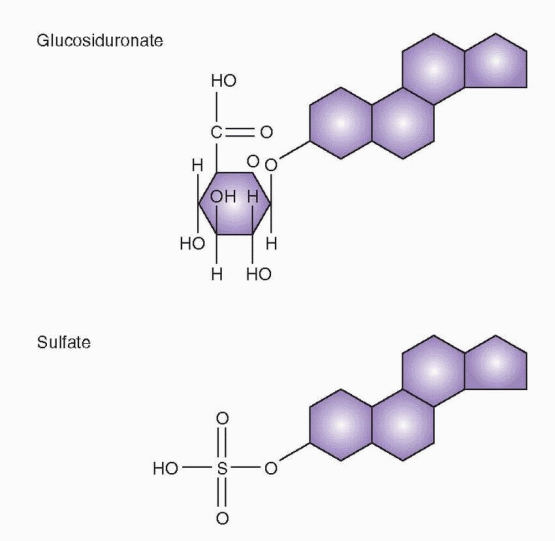
Cells that are capable of clearing estradiol from the circulation accomplish this by biochemical means (conversion to estrone and estriol, moderately effective and very weak estrogens, respectively) and conjugation to products that are water soluble and excreted in the urine and bile (sulfo and glucuro conjugates).
Thus, a steroid hormone has a varied career packed into a short lifetime. In this chapter we will review the important segments of this lifespan in greater detail.
Steroid Hormone Nomenclature
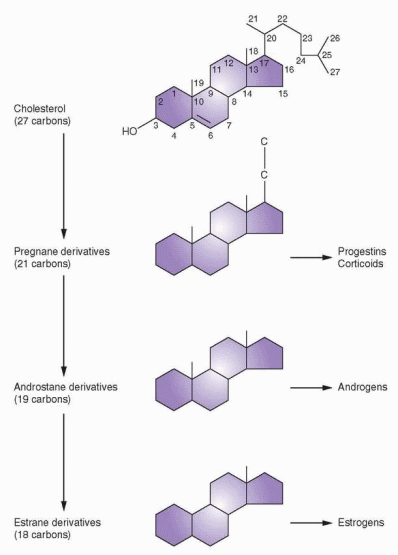
All steroid hormones are of basically similar structure with relatively minor chemical differences leading to striking alterations in biochemical activity. The basic structure is the perhydrocyclopentanephenanthrene molecule. It is composed of three 6-carbon rings and one 5-carbon ring. One 6-carbon ring is benzene, two of the 6-carbon rings are naphthalene, and three 6-carbon rings are phenanthrene; add a cyclopentane (5-carbon ring), and you have the perhydrocyclopentanephenanthrene structure of the steroid nucleus.
The sex steroids are divided into three main groups according to the number of carbon atoms they possess. The 21-carbon series includes the corticoids as well as the progestins, and the basic structure is the pregnane nucleus. The 19-carbon series includes all the androgens and is based on the androstane nucleus, whereas the estrogens are 18-carbon steroids based on the estrane nucleus.
 |
There are six centers of asymmetry on the basic ring structure, and there are 64 possible isomers. Almost all naturally occurring and active steroids are nearly flat, and substituents below and above the plane of the ring are designated alpha (a) (dotted line) and beta (β) (solid line), respectively. Changes in the position of only one substituent can lead to inactive isomers. For example, 17-epitestosterone is considerably weaker than testosterone; the only difference being a hydroxyl group in the a position at C-17 rather than in the β position.
 |
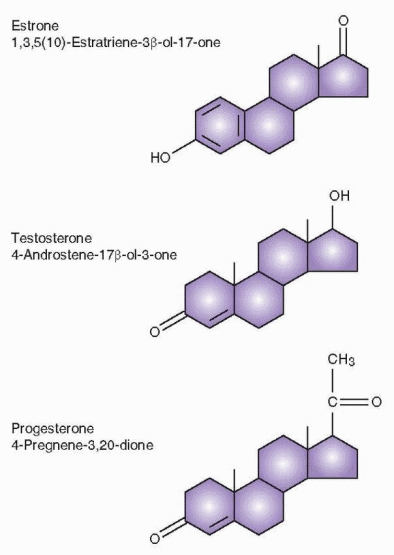
The convention of naming steroids uses the number of carbon atoms to designate the basic name (e.g., pregnane, androstane, or estrane). The basic name is preceded by numbers that indicate the position of double bonds, and the name is altered as follows to indicate one, two, or three double bonds: -ene, -diene, and -triene. Following the basic name, hydroxyl groups are indicated by the number of the carbon attachment, and one, two, or
three hydroxyl groups are designated -ol, -diol, or -triol. Ketone groups are listed last with numbers of carbon attachments, and one, two, or three groups designated -one, -dione, or -trione. Special designations include dehydro, elimination of two hydrogens; deoxy, elimination of oxygen; nor, elimination of carbon; delta or δ, location of double bond.
three hydroxyl groups are designated -ol, -diol, or -triol. Ketone groups are listed last with numbers of carbon attachments, and one, two, or three groups designated -one, -dione, or -trione. Special designations include dehydro, elimination of two hydrogens; deoxy, elimination of oxygen; nor, elimination of carbon; delta or δ, location of double bond.
 |
Lipoproteins and Cholesterol
Cholesterol is the basic building block in steroidogenesis. All steroid-producing organs except the placenta can synthesize cholesterol from acetate. Progestins, androgens, and estrogens, therefore, can be synthesized in situ in the various ovarian tissue compartments from the 2-carbon acetate molecule via cholesterol as the common steroid precursor. However, in situ synthesis cannot meet the demand, and, therefore, the major resource is blood cholesterol that enters the ovarian cells and can be inserted into the biosynthetic pathway or stored in esterified form for later use. The cellular entry of cholesterol is mediated via a cell membrane receptor for low-density lipoprotein (LDL), the bloodstream carrier for cholesterol.
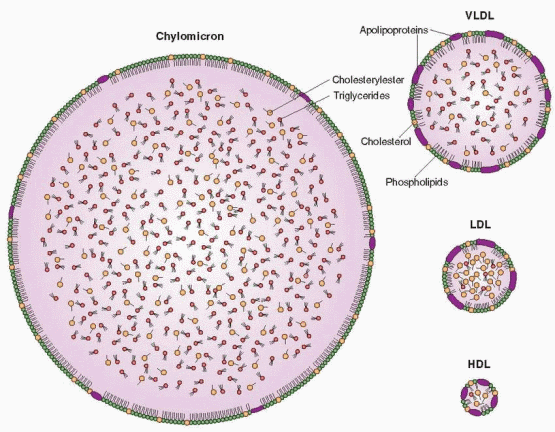
Lipoproteins are large molecules that facilitate the transport of nonpolar fats in a polar solvent, the blood plasma. There are five major categories of lipoproteins according to their charge and density (flotation during ultracentrifugation). They are derived from each other in the following cascade of decreasing size and increasing density.
Chylomicrons
Large, cholesterol (10%)- and triglyceride (90%)-carrying particles formed in the intestine after a fatty meal.
Very Low-Density Lipoproteins (VLDL)
Also carry cholesterol, but mostly triglyceride; more dense than chylomicrons.
Intermediate-Density Lipoproteins (IDL)
Formed (for a transient existence) with the removal of some of the triglyceride from the interior of VLDL particles.
Low-Density Lipoproteins (LDL)
The end products of VLDL catabolism, formed after further removal of triglyceride leaving approximately 50% cholesterol; the major carriers (two-thirds) of cholesterol in the plasma and thus a strong relationship exists between elevated LDL levels and cardiovascular disease.
High-Density Lipoproteins (HDL)
The smallest and most dense of the lipoproteins with the highest protein and phospholipid content; HDL levels are inversely associated with atherosclerosis (high levels are protective). HDL can be further separated into a lighter fraction (HDL2) and a denser fraction (HDL3).
The lipoproteins contain four ingredients: (1) cholesterol in two forms: free cholesterol on the surface of the spherical lipoprotein molecule, and esterified cholesterol in the molecule’s interior; (2) triglycerides in the interior of the sphere; (3) phospholipid; and (4) protein in electrically charged substances on the surface of the sphere and responsible for miscibility with plasma and water. The surface proteins, called apoproteins, constitute the sites that bind to the lipoprotein receptor molecules on the cell surfaces. The principal surface protein of LDL is apoprotein B, and apoprotein A-1 is the principal apoprotein of HDL.
Lipids for peripheral tissues are provided by the secretion of VLDL by the liver. Triglycerides are liberated from VLDL by lipoprotein lipase located in the capillary endothelial cells as well as a lipase enzyme located on the endothelial cells in liver sinusoids. In this process, the surface components (free cholesterol, phospholipids, and apoproteins) are transferred to HDL. Finally, the VLDL is converted to LDL, which plays the important role of transporting cholesterol to cells throughout the body. The hepatic lipase enzyme is sensitive to sex steroid changes: suppression by estrogen and stimulation by androgens.
 |
LDL is removed from the blood by cellular receptors that recognize one of the surface apoproteins. The lipoprotein bound to the cell membrane receptor is internalized and degradated. Intracellular levels of cholesterol are partly regulated by the up- and downregulation of cell membrane LDL receptors. When these LDL receptors are saturated or deficient, LDL is taken up by “scavenger” cells (most likely derived from macrophages) in other tissues, notably the arterial intima. Thus, these cells can become the nidus for atherosclerotic plaques.
HDL is secreted by the liver and intestine or is a product of the degradation of VLDL. Cholesteryl ester molecules move to form a core in a small spherical particle, the HDL3 particle. These particles accept additional free cholesterol, perhaps mediated by receptors that recognize apoprotein A-1. With uptake of cholesterol, the particle size increases to form HDL2, the fraction that reflects changes in diet and hormones. HDL3 levels remain relatively stable.
The protein moieties of the lipoprotein particles are strongly related to the risk of cardiovascular disease, and genetic abnormalities in their synthesis or structure can result in atherogenic conditions. The lipoproteins are a major reason for the disparity in atherosclerosis risk between men and women. Throughout adulthood, the blood HDL-cholesterol level is about 10 mg/dL higher in women, and this difference continues through the postmenopausal years. Total and LDL-cholesterol levels are lower in premenopausal women than in men, but after menopause they rise rapidly.
The protective nature of HDL is due to its ability to pick up free cholesterol from cells or other circulating lipoproteins. This lipid-rich HDL is known as HDL3, which is then converted to the larger, less dense particle, HDL2. Thus, HDL converts lipid-rich scavenger cells (macrophages residing in arterial walls) back to their low-lipid state and carries the excess cholesterol to sites (mainly liver) where it can be metabolized. Another method by which HDL removes cholesterol from the body focuses on the uptake of free cholesterol from cell membranes. The free cholesterol is esterified and moves to the core of the HDL particle. Thus, HDL can remove cholesterol by delivering cholesterol to sites for utilization (steroid-producing cells) or metabolism and excretion (liver).
For good cardiovascular health, the blood concentration of cholesterol must be kept low, and its escape from the bloodstream must be prevented. The problem of cholesterol transport is solved by esterifying the cholesterol and packaging the ester within the cores of plasma lipoproteins. The delivery of cholesterol to cells is in turn solved by lipoprotein receptors. After binding the lipoprotein with its package of esterified cholesterol, the complex is delivered into the cell by receptor-mediated endocytosis (discussed later in this chapter), in which the lysosomes liberate cholesterol for use by the cell.
Major protection against atherosclerosis depends on the high affinity of the receptor for LDL and the ability of the receptor to recycle multiple times, thus allowing large amounts of cholesterol to be delivered while maintaining a healthy low blood level of LDL. Cells can control their uptake of cholesterol by increasing or decreasing the number of LDL receptors according to the intracellular cholesterol levels. Thus, a high-cholesterol diet influences the liver to reduce the number of LDL receptors on its cells, causing an elevated blood level of LDL. Statins protect against atherosclerosis by reducing cholesterol biosynthesis, increasing LDL receptors in the liver, and lowering circulating levels of LDL-cholesterol.
Steroidogenesis
The overall steroid biosynthesis pathway shown in the figure is based primarily on the pioneering work of Kenneth J. Ryan and his coworkers.1,2 These pathways follow a fundamental pattern displayed by all steroid-producing endocrine organs. As a result, it should be no surprise that the normal human ovary produces all three classes of sex steroids: estrogens, progestins, and androgens. The importance of ovarian androgens is appreciated, not only as obligate precursors to estrogens, but also as clinically important secretory products. The ovary differs from the testis in its fundamental complement of critical enzymes and, hence, its distribution of secretory products. The ovary is distinguished from the adrenal gland in that it is deficient in 21-hydroxylase and 11β-hydroxylase reactions. Glucocorticoids and mineralocorticoids, therefore, are not produced in normal ovarian tissue.
During steroidogenesis, the number of carbon atoms in cholesterol or any other steroid molecule can be reduced but never increased. The following reactions can take place:
Cleavage of a side chain (desmolase reaction).
Conversion of hydroxyl groups into ketones or ketones into hydroxyl groups (dehydrogenase reactions).
Addition of OH group (hydroxylation reaction).
Creation of double bonds (removal of hydrogen).
Addition of hydrogen to reduce double bonds (saturation).
The traditional view of steroidogenesis was that each step was mediated by many enzymes, with differences from tissue to tissue. A fundamental simplicity to the system emerged when the responsible complementary DNAs and genes were cloned.3,4 and 5
Steroidogenic enzymes are either dehydrogenases or members of the cytochrome P450 group of oxidases. Cytochrome P450 is a generic term for a family of oxidative enzymes, termed 450 because of a pigment (450) absorbance shift when reduced. P450 enzymes can metabolize many substrates; e.g., in the liver, P450 enzymes metabolize toxins and environmental pollutants. The human genome contains genes for 57 cytochrome P450 enzymes, 7 in mitochondria and 50 in the endoplasmic reticulum (the major site for metabolic clearance). The following distinct P450 enzymes are identified with steroidogenesis: P450scc is the cholesterol side chain cleavage enzyme; P450c11 mediates 11-hydroxylase, 18-hydroxylase, and 19-methyloxidase; P450c17 mediates 17-hydroxylase and 17,20-lyase; P450c21 mediates 21-hydroxylase; and P450arom mediates aromatization of androgens to estrogens. Marked differences in the exonintron organization of the P450 genes are compatible with an ancient origin; thus, the superfamily of P450 genes diverged more than 1.5 billion years ago.
Enzyme | Cellular Location | Reactions |
P450scc | Mitochondria | Cholesterol side chain cleavage |
P450c11 | Mitochondria | 11-hydroxylase 18-hydroxylase 19-methyloxidase |
P450c17 | Endoplasmic reticulum | 17-hydroxylase, 17,20-lyase |
P450c21 | Endoplasmic reticulum | 21-hydroxylase |
P450arom | Endoplasmic reticulum | Aromatase |
The structural knowledge of the P450 enzymes that has been derived from amino acid and nucleotide sequencing studies demonstrated that all the steps between cholesterol and pregnenolone were mediated by a single protein, P450scc, bound to the inner mitochondrial membrane. Cloning data indicate the presence of a single, unique P450scc gene on chromosome 15. These experiments indicated that multiple steps did not require multiple enzymes. Differing activity in different tissues may reflect posttranslational modifications. In addition, these genes contain tissue-specific promoter sequences, which is another reason that regulatory mechanisms can differ in different tissues (e.g., placenta and ovary). P450scc mutations are very rare, producing impaired steroidogenesis in both the adrenal glands and the gonads, causing abnormal phenotypes and adrenal failure.6
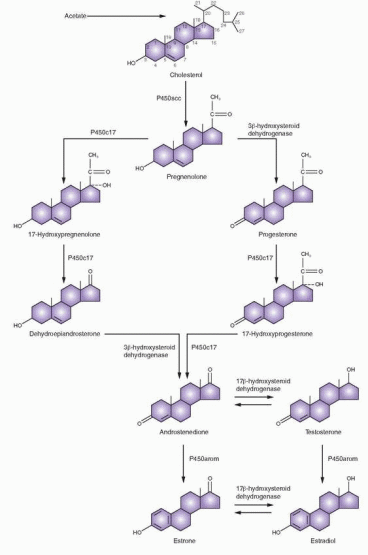
Conversion of cholesterol to pregnenolone involves hydroxylation at the carbon 20 and 22 positions, with subsequent cleavage of the side chain. Conversion of cholesterol to pregnenolone by P450scc takes place within the mitochondria. It is one of the principal effects of tropic hormone stimulation, which also causes the uptake of the cholesterol substrate for this step in the ovary. The tropic hormones from the anterior pituitary bind to the cell surface receptor of the G protein system, activate adenylate cyclase, and increase intracellular cyclic AMP. Cyclic AMP activity leads to gene transcription that encodes the steroidogenic enzymes and accessory proteins. In a process that is faster than gene transcription, cyclic AMP stimulates the hydrolysis of cholesteryl esters and the transport of free cholesterol to the mitochondria.
 |
The cholesterol used for steroid synthesis is derived from circulating low-density lipoproteins (LDL), followed by the mobilization and transport of intracellular stores.5,7,8 LDL cholesterol esters are incorporated into the cell by tropic hormone stimulation of endocytosis via clathrin-coated pits (a mechanism discussed later in this chapter). Cholesterol is stored in the cell in the ester form or as free cholesterol. Indeed, the rate-limiting step in steroidogenesis is the transfer of cholesterol from the outer mitochondrial membrane to the inner mitochondrial membrane where fully active P450scc waits for substrate. The rate-limiting transfer of hydrophobic cholesterol through the aqueous space between the outer and inner mitochondrial membranes is mediated by protein activation stimulated by the tropic hormone. Long-term, chronic steroidogenesis requires gene transcription and protein synthesis, but short-term, acute responses occur independently of new RNA synthesis, although protein synthesis is still necessary, specifically the proteins that regulate cholesterol transfer across the mitochondrial membrane.
Several proteins have been characterized and proposed as regulators of acute intracellular cholesterol transfer. Sterol carrier protein 2 (SCP2) is able to bind and transfer cholesterol between compartments within a cell. Another candidate is a small molecule, steroidogenesis activator polypeptide (SAP), and still another is peripheral benzodiazepine receptor (PBR), which affects cholesterol flux through a pore structure. But the most studied and favored protein as a regulator of acute cholesterol transfer is steroidogenic acute regulator (StAR) protein.9,10,11,12 and 13 StAR messenger RNA and proteins are induced concomitantly with acute steroidogenesis in response to cyclic AMP stimulation. StAR protein increases steroid production and is imported and localized in the mitochondria. But most impressively, congenital lipoid adrenal hyperplasia (an autosomal-recessive disorder) is a failure in adrenal and gonadal steroidogenesis due to a mutation in the StAR gene that results in premature stop codons.14,15 With this mutation, a low level of steroidogenesis is possible, even permitting feminization at puberty, but continuing tropic hormonal stimulation results in an accumulation of intracellular lipid deposits that destroy steroidogenic capability.16 StAR is required for adrenal and gonadal steroidogenesis, and, therefore, is necessary for normal male sexual differentiation.
StAR mediates the transport of cholesterol into mitochondria in adrenal and gonadal steroidogenesis, but not in the placenta and brain. StAR moves cholesterol from the outer mitochondrial membrane to the inner mitochondrial membrane where it can enter the steroidogenic pathway by being converted to prenenolone. A group of proteins structurally related to StAR have been identified, designated StARD4, StARD5, and StARD6. StARD4 is responsible for binding free cholesterol as it is produced in the cytoplasm and transporting it to the outer mitochondrial membrane.12 Because steroid-producing cells do not store large amounts of hormones, acute increases in secretion depend on this system to produce rapid synthesis.
StAR is synthesized in a precursor form as a 285-amino acid protein that has a 25-residue sequence cleaved from the NH2-terminal after transport into mitochondria.17 The mutant forms of StAR undergo premature truncation that prevents this proteolytic cleavage. Mutations of the StAR gene, located on chromosome 8p11.2, are the only inherited disorder of steroidogenesis not caused by a defect in one of the steroidogenic enzymes. The absence of StAR expression in placenta and brain indicates the presence of different mechanisms for cholesterol transport in those tissues.
Once pregnenolone is formed, further steroid synthesis in the ovary can proceed by one of two pathways, either via Δ5-3β-hydroxysteroids or via the Δ4-3-ketone pathway. The first
(the Δ5 pathway) proceeds by way of pregnenolone and dehydroepiandrosterone (DHEA) and the second (the Δ4 pathway) via progesterone and 17a-hydroxyprogesterone.
(the Δ5 pathway) proceeds by way of pregnenolone and dehydroepiandrosterone (DHEA) and the second (the Δ4 pathway) via progesterone and 17a-hydroxyprogesterone.
The conversion of pregnenolone to progesterone involves two steps: the 3β-hydroxysteroid dehydrogenase and Δ4 and 5 isomerase reactions that convert the 3-hydroxyl group to a ketone and transfer the double bond from the 5-6 position to the 4-5 position. The 3β-hydroxysteroid dehydrogenase enzyme catalyzes both the dehydrogenation and isomerization reactions, and exists in two forms (type I and type II), encoded by two separate genes on chromosome 1 (the type I gene is expressed in the placenta, breast, and other non-glandular tissues, the type II gene is expressed in the gonads and the adrenal glands). Once the Δ4 and 5 ketone is formed, progesterone is hydroxylated at the 17 position to form 17a-hydroxyprogesterone. 17a-Hydroxyprogesterone is the immediate precursor of the C-19 (19 carbons) series of androgens in this pathway. By peroxide formation at C-20, followed by epoxidation of the C-17, C-20 carbons, the side chain is split off, forming androstenedione. The 17-ketone may be reduced to a 17β-hydroxyl to form testosterone by the 17β-hydroxysteroid dehydrogenase reaction. Both C-19 steroids (androstenedione and testosterone) can be converted to corresponding C-18 phenolic steroid estrogens (estrone and estradiol) by microsomal reactions in a process referred to as aromatization. This process includes hydroxylation of the angular 19-methyl group, followed by oxidation, loss of the 19-carbon as formaldehyde, and ring A aromatization (dehydrogenation).
As an alternative, pregnenolone can be directly converted to the Δ5-3β-hydroxy C-19 steroid, dehydroepiandrosterone (DHEA), by 17a-hydroxylation followed by cleavage of the side chain. With formation of the Δ4-3-ketone, DHEA is converted into androstenedione. The four reactions involved in converting pregnenolone and progesterone to their 17-hydroxylated products are mediated by a single enzyme, P450c17, bound to smooth endoplasmic reticulum, regulated by a gene on chromosome 10q24.3. 17-Hydroxylase and 17,20-lyase were traditionally regarded as separate enzymes. These two different functions of a single enzyme, P450c17, are not genetic or structural but represent the effect of posttranslational influencing factors.18 In the adrenal gland pathway to cortisol, very little 17,20-lyase activity is expressed. In the ovarian theca cells, the testicular Leydig cells, and the adrenal reticularis, both 17-hydroxylase and 17,20-lyase activities are expressed, directing the steroidogenic pathway via dehydroepiandrosterone (DHEA). In the corpus luteum, the principal pathway is via progesterone.
Characterization of the P450c21 protein and gene cloning indicate that the 21-hydroxylase gene, CYP21, is located on chromosome 6p21.3. An inactive pseudogene, CYP21P, is nearby. Many of the mutations that affect CYP21 and cause congenital adrenal hyperplasia are gene conversions involving recombinations between CYP21 and inactivating mutations in CYP21P.
Aromatization is mediated by P450arom found in the endoplasmic reticulum.19,20 Aromatase cytochrome P450 is derived from chromosome 15q21.1, at a site designated as the CYP19A1 (cytochrome P450, family 19, subfamily A, polypeptide 1) gene, denoting oxidation of the C-19 methyl group. Aromatization in different tissues with different substrates is the result of the single P450arom enzyme encoded by the single gene. A specific haplotype of genetic polymorphisms in CYP19 may be linked to endometrial cancer, a known consequence of excessive estrogenic stimulation of the endometrium.21 Aromatase deficiency because of an inactivating mutation of CYP19A1 is very rare; only a handful of cases have been reported.22 Affected females present at birth with virilization because the placenta cannot convert fetal adrenal androgens to estrogens; thus, maternal virilization during the pregnancy is usually also present.
Aromatase transcription is regulated by several promoter sites that respond to cytokines, cyclic nucleotides, gonadotropins, glucocorticoids, and growth factors.23 Tissue-specific expression is regulated by tissue-specific promoters at the 5′ end of the gene. Thus, this gene has alternative promoters that allow the extremes of highly regulated expression in the
ovary in response to cyclic AMP and gonadotropins, expression in adipose tissue stimulated by prostaglandin E2, and nonregulated expression in the placenta and adipose. Very specific inhibitors of P450arom have been developed, called “aromatase inhibitors,” that allow intense blockage of estrogen production, with clinical applications that include the treatment of breast cancer (e.g., anastrozole and letrozole) and dysfunctional uterine bleeding. The aromatase complex also includes NADPH-cytochrome P450 reductase, a ubiquitous flavoprotein involved in reduction reactions.
ovary in response to cyclic AMP and gonadotropins, expression in adipose tissue stimulated by prostaglandin E2, and nonregulated expression in the placenta and adipose. Very specific inhibitors of P450arom have been developed, called “aromatase inhibitors,” that allow intense blockage of estrogen production, with clinical applications that include the treatment of breast cancer (e.g., anastrozole and letrozole) and dysfunctional uterine bleeding. The aromatase complex also includes NADPH-cytochrome P450 reductase, a ubiquitous flavoprotein involved in reduction reactions.
The 17β-hydroxysteroid dehydrogenase and 5a-reductase reactions are mediated by non-P450 enzymes. The 17β-hydroxysteroid dehydrogenase is bound to the endoplasmic reticulum and the 5a-reductase to the nuclear membrane. The 17β-hydroxysteroid dehydrogenase enzymes convert estrone to estradiol, androstenedione to testosterone, and DHEA to androstenediol, and vice versa. Eight different isozymes have been cloned and characterized.24 The type 1 enzyme is active in the placenta and granulosa cells, converting estrone to estradiol. The type 2 and 4 enzymes, found in many tissues, form androstenedione and estrone from testosterone and estradiol, respectively. The type 3 and 5 enzymes in the testis reduce androstenedione to testosterone. The type 6 enzyme may be found only in rodents, and the type 7 and 8 enzymes are widespread, but have limited activity. Thus types 1,3, and 5 form active estrogens and androgens, whereas types 2 and 4 produce weaker products, a form of inactivation, important, for example, in protecting the fetus against testosterone and estradiol in the maternal circulation. The cell-specific production of each of these isoforms is a method for regulating the local concentration of estrogens and androgens.
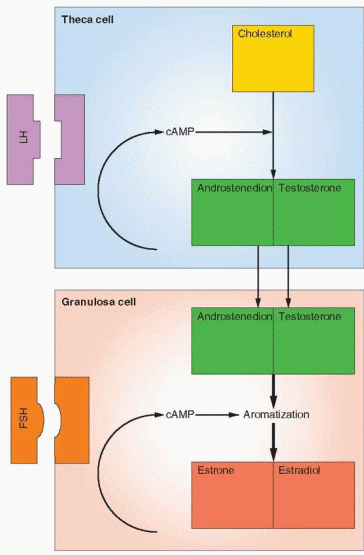
The Two-Cell System
The two-cell system is a logical explanation of the events involved in ovarian follicular steroidogenesis.25 This explanation, first proposed by Falck in 1959,26 brings together information on the site of specific steroid production, along with the appearance and importance of hormone receptors. The following facts are important:
FSH receptors are present on the granulosa cells.
FSH receptors are induced by FSH itself.
LH receptors are present on the theca cells and initially absent on the granulosa cells, but, as the follicle grows, FSH induces the appearance of LH receptors on the granulosa cells.
FSH induces aromatase enzyme activity in granulosa cells.
The above actions are modulated by autocrine and paracrine factors secreted by the theca and granulosa cells.
These facts combine into the two-cell system to explain the sequence of events in ovarian follicular growth and steroidogenesis. The initial change from a primordial follicle to a preantral follicle is independent of hormones, and the stimulus governing this initial step in growth is unknown. Continued growth, however, depends on FSH stimulation. As the granulosa responds to FSH, proliferation and growth are associated with an increase in FSH receptors, a specific effect of FSH itself, but an action that is enhanced very significantly by the autocrine and paracrine peptides. The theca cells are characterized by steroidogenic activity in response to LH, specifically resulting in androgen production, by transcription of the P450scc, P450c17, and 3β-hydroxysteroid dehydrogenase genes. Aromatization of
androgens to estrogens is a distinct activity within the granulosa layer induced by FSH by activation of the P450arom gene. Androgens produced in the theca layer, therefore, must diffuse into the granulosa layer. In the granulosa layer they are converted to estrogens, and the increasing level of estradiol in the peripheral circulation reflects release of the estrogen back toward the theca layer and into blood vessels.
androgens to estrogens is a distinct activity within the granulosa layer induced by FSH by activation of the P450arom gene. Androgens produced in the theca layer, therefore, must diffuse into the granulosa layer. In the granulosa layer they are converted to estrogens, and the increasing level of estradiol in the peripheral circulation reflects release of the estrogen back toward the theca layer and into blood vessels.
The theca and granulosa cells secrete peptides that operate as both autocrine and paracrine factors.27 Insulin-like growth factor (IGF) is secreted by the theca and enhances the LH stimulation of androgen production in the theca cells as well as FSH-mediated aromatization in the granulosa. Evidence indicates that the endogenous insulin-like growth factor in the human ovarian follicle is IGF-II in both the granulosa and the theca cells.28 Studies indicating activity of IGF-I with human ovarian tissue can be explained by the fact that both IGF-I and IGF-II activities can be mediated by the type I IGF receptor, which is structurally similar to the insulin receptor. The regulation of FSH receptors on granulosa cells is relatively complex. Although FSH increases the activity of its own receptor gene in a cyclic AMP-mediated mechanism, this action is influenced by inhibitory agents, such as epidermal growth factor, fibroblast growth factor, and even a gonadotropin-releasing hormone (GnRH)-like protein. Inhibin and activin are produced in the granulosa in response to FSH, and activin has the important autocrine role of enhancing FSH actions, especially the production of FSH receptors. Inhibin enhances LH stimulation of androgen synthesis in the
theca to provide a substrate for aromatization to estrogen in the granulosa, whereas activin suppresses androgen synthesis. This important paracrine regulation of androgen production in thecal cells by inhibin and activin, discussed in Chapter 6, is exerted primarily through modification of the expression of steroidogenic enzymes, especially P450c17.29
theca to provide a substrate for aromatization to estrogen in the granulosa, whereas activin suppresses androgen synthesis. This important paracrine regulation of androgen production in thecal cells by inhibin and activin, discussed in Chapter 6, is exerted primarily through modification of the expression of steroidogenic enzymes, especially P450c17.29
 |
After ovulation, the dominance of the luteinized granulosa layer is dependent on preovulatory induction of an adequate number of LH receptors, and, therefore, dependent on adequate FSH action. Prior to ovulation the granulosa layer is characterized by aromatization activity and conversion of the theca androgens to estrogens, an FSH-mediated activity. After ovulation the granulosa layer secretes progesterone and estrogens directly into the bloodstream, an LH-mediated activity.
Granulosa and theca cells each have an androgen aromatase system that can be demonstrated in vitro. However, in vivo, the activity of the granulosa layer in the follicular phase is several hundred times greater than the activity of the theca layer, and, therefore, the granulosa is the main biosynthetic source of estrogen in the growing follicle.30 Because granulosa cells lack P450c17, the rate of aromatization in the granulosa layer is directly related to and dependent on the androgen substrate made available by the theca cells. Hence, estrogen secretion by the follicle prior to ovulation is the result of combined LH and FSH stimulation of the two cell types, the theca and the granulosa. After ovulation, it is believed the two cell types continue to function as a two-cell system; luteal cells derived from theca produce androgens for aromatization into estrogens by luteal cells derived from granulosa.
Free (Unbound (%)) | Albumin-Bound (%) | SHBC-Bound (%) | |
Estrogen | 1 | 30 | 69 |
Testosterone | 1 | 30 | 69 |
DHEA | 4 | 88 | 8 |
Androstenedione | 7 | 85 | 8 |
Dihydrotestosterone | 1 | 71 | 28 |
From Mendel31 | |||
Blood Transport of Steroids
While circulating in the blood, a majority of the principal sex steroids, estradiol and testosterone, is bound to a protein carrier, known as sex hormone-binding globulin (SHBG) produced mainly in the liver. Another 30% is loosely bound to albumin, leaving only about 1% unbound and free. A very small percentage also binds to corticosteroid-binding globulin. Hyperthyroidism, pregnancy, and estrogen administration all increase SHBG levels, whereas corticoids, androgens, progestins, growth hormone, insulin, and IGF-I decrease SHBG.
The circulating level of SHBG is inversely related to body weight, and, thus, significant weight gain can decrease SHBG and produce important changes in the unbound levels of the sex steroids. Another important mechanism for a reduction in circulating SHBG levels is insulin resistance and hyperinsulinemia.32,33 Thus, increased insulin levels in the circulation lower SHBG levels, and this may be the major mechanism that mediates the impact of increased body weight on SHBG. This relationship between the levels of insulin and SHBG is so strong that SHBG concentrations are a marker for hyperinsulinemic insulin resistance, and a low level of SHBG is a predictor for the development of type 2 diabetes mellitus.34
The distribution of body fat has a strong influence on SHBG levels. Android or central fat is located in the abdominal wall and visceral-mesenteric locations. This fat distribution
is associated with hyperinsulinemia, hyperandrogenism, and decreased levels of SHBG.35 The common mechanism for these changes is probably the hyperinsulinemia.
is associated with hyperinsulinemia, hyperandrogenism, and decreased levels of SHBG.35 The common mechanism for these changes is probably the hyperinsulinemia.
SHBG is a glycoprotein that contains a single binding site for androgens and estrogens, even though it is a homodimer composed of two monomers. Its gene has been localized to the short arm (p12-13) of chromosome 17.36 Genetic studies have revealed that the SHBG gene also encodes the androgen-binding protein present in the seminiferous tubules, synthesized by the Sertoli cells.37,38 Dimerization is believed to be necessary to form the single steroidbinding site. Specific chromosomal abnormalities with decreased or abnormal SHBG have not been reported. SHBG gene expression has now been identified in other tissues (brain, placenta, and endometrium), although a biologic significance has not been determined.
Transcortin, also called corticosteroid-binding globulin, is a plasma glycoprotein that binds cortisol, progesterone, deoxycorticosterone, corticosterone, and some of the other minor corticoid compounds. Normally about 75% of circulating cortisol is bound to transcortin, 15% is loosely bound to albumin, and 10% is unbound or free. Progesterone circulates in the following percentages: less than 2% unbound, 80% bound to albumin, 18% bound to transcortin, and less than 1% bound to SHBG. Binding in the circulation follows the law of mass action: the amount of the free, unbound hormone is in equilibrium with the bound hormone. Thus, the total binding capacity of a binding globulin will influence the amount that is free and unbound.
The biologic effects of the major sex steroids are largely determined by the unbound portion, known as the free hormone. In other words, the active hormone is unbound and free, whereas the bound hormone is relatively inactive. This concept is not without controversy. The hormone-protein complex may be involved in an active uptake process at the target cell plasma membrane.39,40 and 41 The albumin-bound fraction of steroids may also be available for cellular action because this binding has low affinity. Because the concentration of albumin in plasma is manyfold greater than that of SHBG, the contribution of the albumin-bound fraction can be significant. Routine assays determine the total hormone concentration, bound plus free, and special steps are required to measure the active free level of testosterone, estradiol, and cortisol.
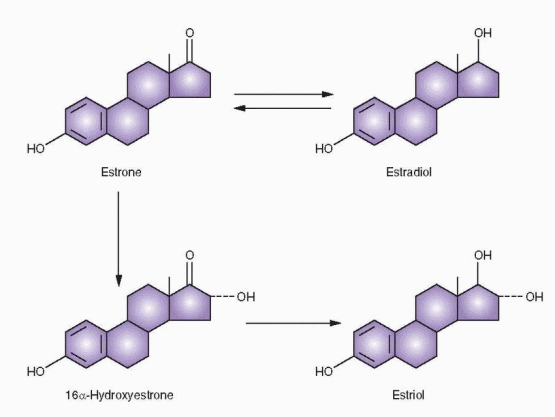
Estrogen Metabolism
Androgens are the precursors of estrogens. 17β-Hydroxysteroid dehydrogenase activity converts androstenedione to testosterone, which is not a major secretory product of the normal ovary. It is rapidly demethylated at the C-19 position and aromatized to estradiol, the major estrogen secreted by the human ovary. Estradiol also arises to a major degree from androstenedione via estrone, and estrone itself is secreted in significant daily amounts. Estriol is the peripheral metabolite of estrone and estradiol and not a secretory product of the ovary. The formation of estriol is typical of general metabolic “detoxification,” conversion of biologically active material to less active forms.
The conversion of steroids in peripheral tissues is not always a form of inactivation. Free androgens are peripherally converted to free estrogens, for example, in skin and adipose cells. The location of the adipose cells influences their activity. Women with central obesity (the abdominal area) produce more androgens.42 The work of Siiteri and MacDonald43 demonstrated that enough estrogen can be derived from circulating androgens to produce bleeding in the postmenopausal woman. In the female the adrenal gland remains the major source of circulating androgens, in particular androstenedione. In the male, almost all of the circulating estrogens are derived from peripheral conversion of androgens. The precursor
androgens consist principally of androstenedione, dehydroepiandrosterone, and dehydroepiandrosterone sulfate.
androgens consist principally of androstenedione, dehydroepiandrosterone, and dehydroepiandrosterone sulfate.
 |
It can be seen, therefore, that the pattern of circulating steroids in the female is influenced by the activity of various processes outside the ovary. Because of the peripheral contribution to steroid levels, the term secretion rate is reserved for direct organ secretion, whereas production rate includes organ secretion plus peripheral contribution via conversion of precursors. The metabolic clearance rate (MCR) equals the volume of blood that is cleared of the hormone per unit of time. The blood production rate (PR) then equals the metabolic clearance rate multiplied by the concentration of the hormone in the blood.
MCR = Liters/Day
Pr = MCR × Concentration (Liters/Day × Amount/Liter = Amount/Day)
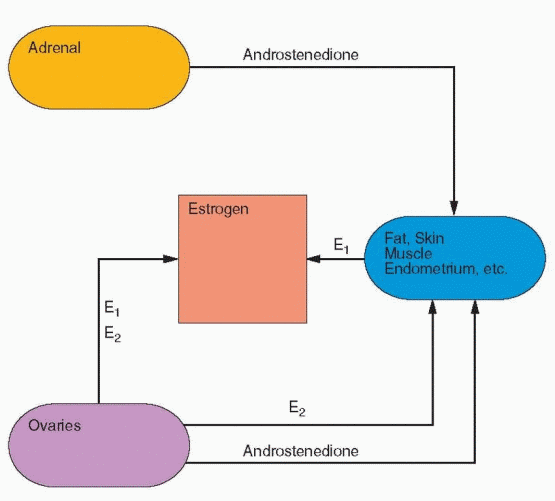 |
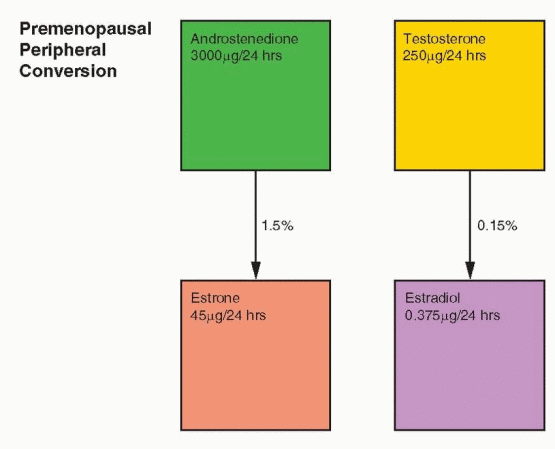
In the normal nonpregnant female, estradiol is produced at the rate of 100-300 μg/day. The production of androstenedione is about 3 mg/day, and the peripheral conversion (about 1%) of androstenedione to estrone accounts for about 20-30% of the estrone produced per day. Because androstenedione is secreted in milligram amounts, even a small percent conversion to estrogen results in a significant contribution to estrogens, which exist and function in the circulation in picogram amounts. Thus, the circulating estrogens in the female are the sum of direct ovarian secretion of estradiol and estrone, plus peripheral conversion of C-19 precursors. Whereas estradiol is produced in μg amounts, it circulates and functions within cells in concentrations of pg/mL.
 |
Progesterone Metabolism
Peripheral conversion of steroids to progesterone is not seen in the nonpregnant female; rather, the progesterone production rate is a combination of secretion from the adrenal and the ovaries. Including the small contribution from the adrenal, the blood production rate of progesterone in the preovulatory phase is less than 1 mg/day. During the luteal phase, production increases to 20-30 mg/day. The metabolic fate of progesterone, as expressed by its many excretion products, is more complex than estrogen. About 10-20% of progesterone is excreted as pregnanediol.
Pregnanediol glucuronide is present in the urine in concentrations less than 1 mg/day until ovulation. Postovulation pregnanediol excretion reaches a peak of 3-6 mg/day, which is maintained until 2 days prior to menses. The assay of pregnanediol in the urine now has little clinical use.
In the preovulatory phase in adult females, in all prepubertal females, and in the normal male, the blood levels of progesterone are at the lower limits of immunoassay sensitivity: less than 1 ng/mL. After ovulation, i.e., during the luteal phase, progesterone ranges from 3 to 15 ng/mL. In congenital adrenal hyperplasia, progesterone blood levels can be as high as 50 times above normal.
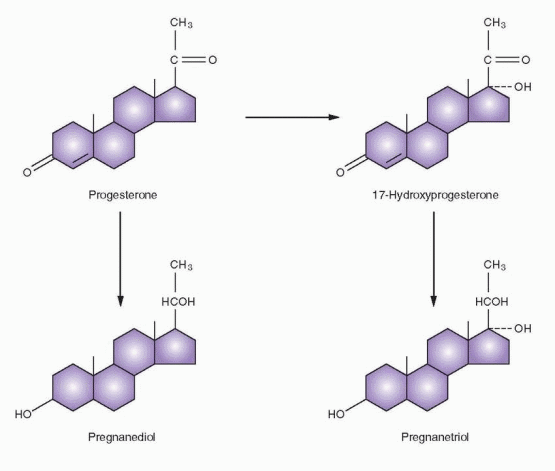 |
Pregnanetriol is the chief urinary metabolite of 17a-hydroxyprogesterone and has clinical significance in the adrenogenital syndrome, a syndrome of virilizing adrenal hyperplasia in which an enzyme defect results in accumulation of 17a-hydroxyprogesterone and increased excretion of pregnanetriol. The inherited abnormality in virilizing adrenal hyperplasia results in an inability to synthesize glucocorticoids. The hypothalamic-pituitary axis reacts to the low level of cortisol by elevated ACTH secretion in a homeostatic response to achieve normal levels of cortisol production. This stimulation induces a hyperplastic adrenal cortex that produces androgens as well as corticoid precursors in abnormal quantities. The plasma or serum assay of 17a-hydroxyprogesterone is a more sensitive and accurate index of this enzyme deficiency than measurement of pregnanetriol. Normally, the blood level of 17a-hydroxyprogesterone is less than 100 ng/dL, although after ovulation and during the luteal phase of a normal menstrual cycle, a peak of 200 ng/dL can be reached. In syndromes of adrenal hyperplasia, values can be 10-400 times normal.
Androgen Metabolism
The major androgen products of the ovary are dehydroepiandrosterone (DHEA) and androstenedione (and only a little testosterone), which are secreted mainly by stromal tissue derived from theca cells. With excessive accumulation of stromal tissue or in the presence of an androgen-producing tumor, testosterone becomes a significant secretory product. Occasionally, a nonfunctioning tumor can induce stromal proliferation and increased androgen production. The normal accumulation of stromal tissue at midcycle
results in a rise in circulating levels of androstenedione and testosterone at the time of ovulation.
results in a rise in circulating levels of androstenedione and testosterone at the time of ovulation.
The adrenal cortex produces three groups of steroid hormones: the glucocorticoids, the mineralocorticoids, and the sex steroids. The adrenal sex steroids represent intermediate byproducts in the synthesis of glucocorticoids and mineralocorticoids, and excessive secretion of the sex steroids occurs only with neoplastic cells or in association with enzyme deficiencies. Under normal circumstances, adrenal gland production of the sex steroids is less significant than gonadal production of androgens and estrogens. About one-half of the daily production of DHEA and androstenedione comes from the adrenal gland; the other half of androstenedione is secreted by the ovary, but the other half of DHEA is split almost equally between the ovary and peripheral tissues. The production rate of testosterone in the normal female is 0.2-0.3 mg/day, and approximately 50% arises from peripheral conversion of androstenedione (and a small amount from DHEA) to testosterone, whereas 25% is secreted by the ovary and 25% by the adrenal. The major androgens are excreted in the urine as 17-ketosteroids.
There is no circadian cycle of the major sex steroids in the female. However, short-term variations in the blood levels due to episodic secretion require multiple sampling for absolutely accurate assessment. Although frequent sampling is necessary for a high degree of accuracy, a random sample is sufficient for clinical purposes to determine whether a level is within a normal range.
The testosterone-binding capacity is decreased by androgens; hence, the binding capacity in men is lower than that in normal women. The binding globulin level in women with increased androgen production is also depressed. Androgenic effects are dependent on the unbound fraction that can move freely from the vascular compartment into the target cells. Routine assays determine the total hormone concentration, bound plus free. Thus, a total testosterone concentration can be in the normal range in a woman who is hirsute or even virilized, but because the binding globulin level is depressed by the androgen effects, the percent free and active testosterone is elevated. The need for a specific assay for the free portion of testosterone can be questioned because the very presence of hirsutism or virilism indicates increased androgen effects. In the face of hirsutism, one can reliably interpret a normal testosterone level as compatible with decreased binding capacity and increased active free testosterone.
Both total and unbound testosterone are normal in only a few women with hirsutism. In these cases, the hirsutism, heretofore regarded as idiopathic, most likely results from excessive intracellular androgen effects (specifically increased intracellular conversion of testosterone to dihydrotestosterone).
Reduction of the Δ4 unsaturation (an irreversible pathway) in testosterone is very significant, producing derivatives very different in their spatial configuration and activity. The 5β-derivatives are not androgenic, and this is not an important pathway; however, the 5a-derivative (a very active pathway) is extremely potent. Indeed, dihydrotestosterone (DHT), the 5a-derivative, is the principal androgenic hormone in a variety of target tissues and is formed within the target tissue itself.
In men, the majority of circulating DHT is derived from testosterone that enters a target cell and is converted by means of 5a-reductase to DHT. In women, because the production rate of androstenedione is greater than testosterone, blood DHT is primarily derived from androstenedione and partly from dehydroepiandrosterone.44 Thus, in women, the skin production of DHT is predominantly influenced by androstenedione. DHT is by definition an intracrine hormone, formed and acting within target tissues.45 The 5a-reductase enzyme exists in two forms, type I and II, each encoded by a separate gene, with the type I enzyme found in skin and the type II reductase predominantly expressed in reproductive tissues.46
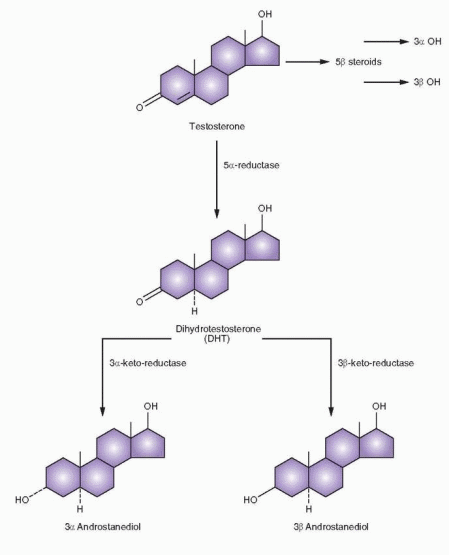 |
DHT is largely metabolized intracellularly; hence, the blood DHT is only about one-tenth the level of circulating testosterone, and it is clear that testosterone is the major circulating androgen. In tissues sensitive to DHT (which includes hair follicles), only DHT enters the nucleus to provide the androgen message. DHT also can perform androgenic actions within cells that do not possess the ability to convert testosterone to DHT. DHT is further reduced by a 3a-keto-reductase to androstanediol, which is relatively inactive. The metabolite of androstanediol, 3a-androstanediol glucuronide, is the major metabolite of DHT and can be measured in the plasma, indicating the level of activity of target tissue conversion of testosterone to DHT.
Not all androgen-sensitive tissues require the prior conversion of testosterone to DHT. In the process of masculine differentiation, the development of the wolffian duct structures (epididymis, the vas deferens, and the seminal vesicle) is dependent on testosterone as the intracellular mediator, whereas development of the urogenital sinus and urogenital tubercle into the male external genitalia, urethra, and prostate requires the conversion of testosterone to DHT.47 Muscle development is under the direct control of testosterone. Testosterone is also aromatized to a significant extent in the brain, liver, and breast; and in some circumstances (e.g., in the brain) androgenic messages can be transmitted via estrogen.
The Importance of Local Sex Hormone Production
The characteristics of sex steroid metabolism reviewed above contribute to an important clinical concept: circulating levels of sex hormones do not always reflect concentrations in target cells. In premenopausal women, target tissues synthesize and metabolize most of the testosterone produced. Thus, in women testosterone functions as a paracrine and intracrine hormone. In men, abundant secretion of testosterone creates circulating levels that are sufficient to allow testosterone to function as a classic hormone. In women, the same description applies to estradiol. Estradiol functions as a classical circulating hormone until menopause, after which both estradiol and testosterone activities are due to local target tissue synthesis, using precursors derived from the circulation. Clinical interventions after menopause, therefore, are directed to local hormone production; for example, the use of aromatase inhibitors to treat breast cancer.
Excretion of Steroids
Active steroids and metabolites are excreted as sulfo and glucuro conjugates. Conjugation of a steroid converts a hydrophobic compound into a hydrophilic one and generally reduces or eliminates the activity of a steroid. This is not completely true, however, because hydrolysis of the ester linkage can occur in target tissues and restore the active form. Furthermore, estrogen conjugates can have biologic activity, and it is known that sulfated conjugates are actively secreted and may serve as precursors, present in the circulation in relatively high concentrations because of binding to serum proteins. Ordinarily, however, conjugation by liver and intestinal mucosa is a step in deactivation preliminary to, and essential for, excretion into urine and bile.
 |
Cellular Mechanism of Action
Hormones circulate in extremely low concentrations and, in order to respond with specific and effective actions, target cells require the presence of special mechanisms. There are two major types of hormone action at target tissues. One mediates the action of tropic hormones (peptide and glycoprotein hormones) with receptors at the cell membrane level. In contrast, the smaller steroid hormones enter cells readily, and the basic mechanism of action involves specific receptor molecules within the cells. It is the affinity, specificity, and activity of the receptors, together with the large concentration of receptors in cells, that allow a small amount of hormone to produce a biologic response.
The many different types of receptors can be organized into the following basic categories:
Intracellular Receptors
Receptors within cells lead to transcription activation. Examples include the receptors for estrogen and thyroid hormones.
G Protein Receptors
These receptors are composed of a single polypeptide chain that spans the cell membrane. Binding to a specific hormone leads to interaction with G proteins that, in turn, activate second messengers. Examples include receptors for tropic hormones, prostaglandins, light, and odors. The second messengers include the adenylate cyclase enzyme, the phospholipase system, and calcium ion changes.
Ion Gate Channels
These cell surface receptors are composed of multiple units, that after binding, open ion channels. The influx of ions changes the electrical activity of the cells. The best example of this type is the acetylcholine receptor.
Receptors with Intrinsic Enzyme Activity
These transmembrane receptors have an intracellular component with tyrosine or serine kinase activity. Binding leads to receptor autophosphorylation and activity. Examples include the receptors for insulin and growth factors (tyrosine kinase) and the receptors for activin and inhibin (serine kinase).
The System of Internalization
Receptors that do not fit the above categories include the receptors for LDL, prolactin, growth hormone, and some of the growth factors. These receptors allow entry of their ligands into cells by the process of endocytosis (discussed later in this chapter).
Mechanism of Action for Steroid Hormones
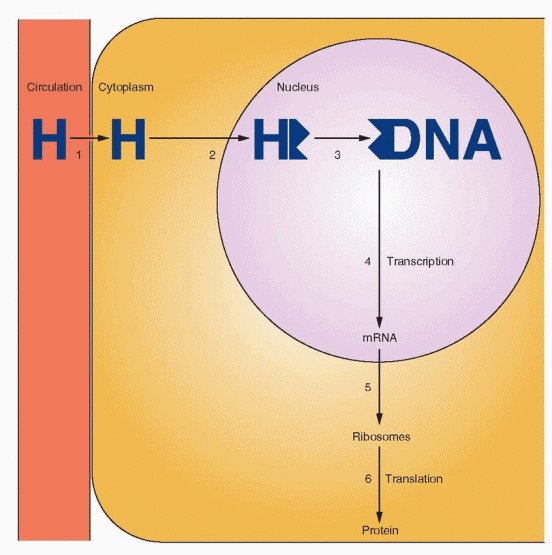
The specificity of the reaction of tissues to sex steroid hormones is due to the presence of intracellular receptor proteins. Different types of tissues, such as liver, kidney, and uterus, respond in a similar manner. The mechanism includes: (1) steroid hormone diffusion across the cell membrane, (2) steroid hormone binding to a receptor protein, (3) interaction of a hormone-receptor complex with nuclear DNA, (4) synthesis of messenger RNA (mRNA), (5) transport of the mRNA to the ribosomes, and finally, (6) protein synthesis in the cytoplasm that results in specific cellular activity. The steroid hormone receptors primarily affect gene transcription, but also regulate posttranscriptional events and nongenomic events. Steroid receptors regulate gene transcription through multiple mechanisms, not all of which require direct interactions with DNA.
 |
Each of the major classes of the sex steroid hormones, including estrogens, progestins, and androgens, act according to this general mechanism. Glucocorticoid and mineralocorticoid receptors, when in the unbound state, reside in the cytoplasm and move into the nucleus after hormone-receptor binding. Estrogens, progestins, and androgens are transferred across the nuclear membrane and bind to their receptors within the nucleus.
Steroid hormones are rapidly transported across the cell membrane by simple diffusion. The factors responsible for this transfer are unknown, but the concentration of free (unbound) hormone in the bloodstream seems to be an important and influential determinant of cellular function. Once in the cell, the sex steroid hormones bind to their individual receptors. During this process, transformation or activation of the receptor occurs. Transformation refers to a conformational change of the hormone-receptor complex revealing or producing a binding site that is necessary in order for the complex to bind to the chromatin. In the unbound state, the receptor is associated with heat shock proteins that stabilize and protect the receptor and maintain a conformational shape that keeps the DNA binding region in an inactive state. Activation of the receptor is driven by hormone binding that causes a dissociation of the receptor-heat shock protein complex.
The hormone-receptor complex binds to specific DNA sites (hormone-responsive elements) that are located upstream of the gene. The specific binding of the hormone-receptor complex with DNA results in RNA polymerase initiation of transcription. Transcription leads to translation, mRNA-mediated protein synthesis on the ribosomes. The principal action of steroid hormones is the regulation of intracellular protein synthesis by means of the receptor mechanism.
Biologic activity is maintained only while the nuclear site is occupied with the hormonereceptor complex. The dissociation rate of the hormone and its receptor as well as the halflife of the nuclear chromatin-bound complex are factors in the biologic response because the hormone response elements are abundant and, under normal conditions, are occupied only to a small extent.48 Thus, an important clinical principle is the following: duration of exposure to a hormone is as important as dose. One reason only small amounts of estrogen need be present in the circulation is the long half-life of the estrogen hormone-receptor complex. Indeed, a major factor in the potency differences among the various estrogens (estradiol, estrone, estriol) is the length of time the estrogen-receptor complex occupies the nucleus. The higher rate of dissociation with the weak estrogen (estriol) can be compensated for by continuous application to allow prolonged nuclear binding and activity. Cortisol and progesterone must circulate in large concentrations because their receptor complexes have short half-lives in the nucleus.
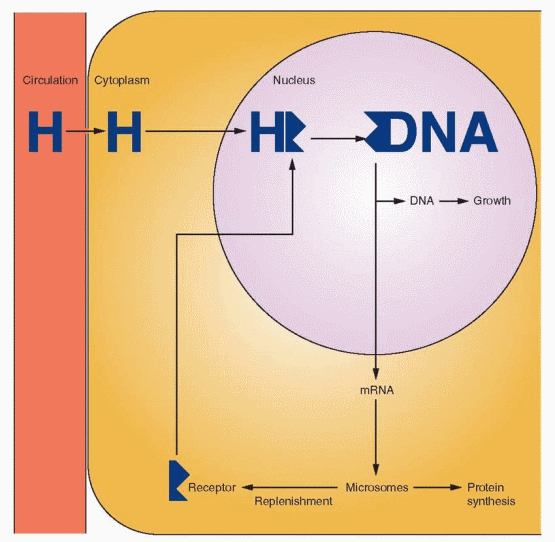
An important action of estrogen is the modification of its own and other steroid hormone activity by affecting receptor concentrations. Estrogen increases target tissue responsiveness to itself and to progestins and androgens by increasing the concentration of its own receptor and that of the intracellular progestin and androgen receptors. Progesterone and clomiphene, on the other hand, limit tissue response to estrogen by blocking this mechanism, thus decreasing over time the concentration of estrogen receptors. Small amounts of receptor depletion and small amounts of steroid in the blood activate the mechanism.
The synthesis of the sex steroid receptors obviously takes place in the cytoplasm, but with estrogen and progestin receptors, synthesis must be quickly followed by transportation into the nucleus. There is an amazingly extensive nuclear traffic.49 The nuclear membrane contains 3,000 to 4,000 pores. A cell synthesizing DNA imports about one million histone molecules from the cytoplasm every 3 minutes. If the cell is growing rapidly, about three newly assembled ribosomes will be transported every minute in the other direction. The typical cell can synthesize 10,000 to 20,000 different proteins. How do they know where to go? The answer is that these proteins have localization signals. In the case of steroid hormone receptor proteins, the signal sequences are in the hinge region.
 |
Estrogen and progestin receptors exit continuously from the nucleus to the cytoplasm and are actively transported back to the nucleus. This is a constant shuttle; diffusion into the cytoplasm is balanced by the active transport into the nucleus. This raises the possibility that some diseases are due to poor traffic control. This can be true of some acquired diseases as well, e.g., Reye’s syndrome, an acquired disorder of mitochondrial enzyme function.
The fate of the hormone-receptor complex after gene activation is referred to as hormone-receptor processing. In the case of estrogen receptors, processing involves the rapid degradation of receptors unbound with estrogen, and a much slower degradation of bound receptors after gene transcription. The rapid turnover of estrogen receptors has clinical significance. The continuous presence of estrogen is an important factor for continuing response.
The best example of the importance of these factors is the difference between estradiol and estriol. Estriol has only 20-30% affinity for the estrogen receptor compared with estradiol; therefore, it is rapidly cleared from a cell. But if the effective concentration is kept equivalent to that of estradiol, it can produce a similar biologic response.50 In pregnancy, where the concentration of estriol is very great, it can be an important hormone, not just a metabolite.
The depletion of estrogen receptors in the endometrium by progestational agents is the fundamental reason for adding progestins to estrogen treatment programs. The progestins
accelerate the turnover of preexisting receptors, and this is followed by inhibition of estrogen-induced receptor synthesis. Using monoclonal antibody immunocytochemistry, this action has been pinpointed to the interruption of transcription in estrogen-regulated genes. The mechanism is different for androgen antiestrogen effects. Androgens also decrease estrogen receptors within target tissues, especially in the uterus.51,52
accelerate the turnover of preexisting receptors, and this is followed by inhibition of estrogen-induced receptor synthesis. Using monoclonal antibody immunocytochemistry, this action has been pinpointed to the interruption of transcription in estrogen-regulated genes. The mechanism is different for androgen antiestrogen effects. Androgens also decrease estrogen receptors within target tissues, especially in the uterus.51,52
The Receptor Superfamily
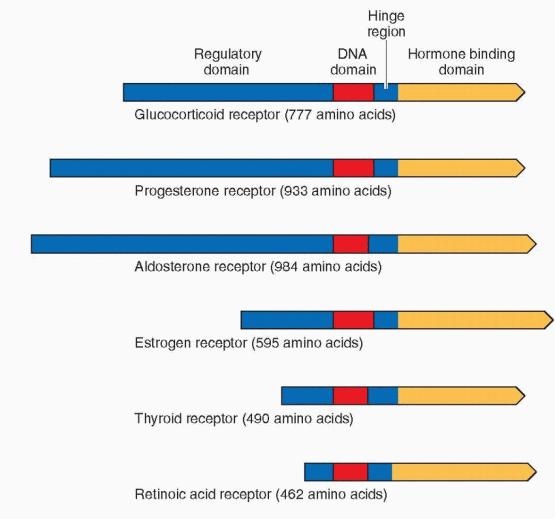
Recombinant DNA techniques have permitted the study of the gene sequences that code for the synthesis of nuclear receptors. Steroid hormone receptors share a common structure with the receptors for thyroid hormone, 1,25-dihydroxyvitamin D3, and retinoic acid; thus, these receptors are called a superfamily.53,54 Each receptor contains characteristic domains that are similar and interchangeable. Therefore, it is not surprising that the specific hormones can interact with more than one receptor in this family. Analysis of these receptors suggests a complex evolutionary history during which gene duplication and swapping between domains of different origins occurred. This family now includes hundreds of proteins, present in practically all species, from worms to insects to humans. Some are called orphan receptors because specific ligands for these proteins have not been identified, but the number of orphan receptors is gradually diminishing (deorphaning). It has been convincingly argued that the 6 steroid receptors originated in a common ancestral receptor gene.55 The identification of steroid receptors in the sea lamprey dates the origin to over 450 million years ago, and the characterization of a receptor that functions like an estrogen receptor in the mollusk indicates that the ancient and initial sex steroid receptor was an estrogen receptor.56 Knowledge of the complete human genome has confirmed that there are 48 nuclear receptors in the receptor superfamily.54
 |
The Estrogen Receptors
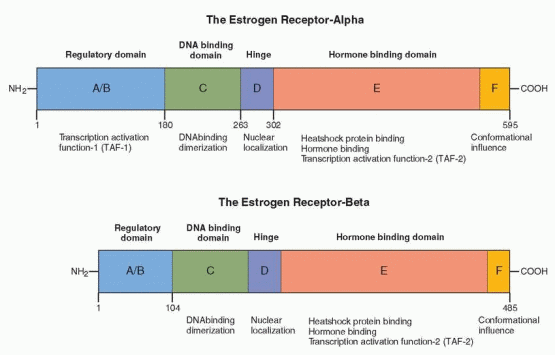
Two estrogen receptors have been identified, designated as estrogen receptor-alpha (ER-a) and estrogen receptor-beta (ER-β).57,58 The estrogen receptor-a was discovered about 1960, and the amino acid sequence was reported in 1986.59,60 and 61 The estrogen receptor-a is translated from a 6.8-kilobase mRNA derived from a gene that contains eight exons on the long arm of chromosome 6.62 It has a molecular weight of approximately 66,000 with 595 amino acids. The receptor-a half-life is approximately 4-7 hours; thus the estrogen receptor-a is a protein with a rapid turnover. The more recently discovered estrogen receptor-β, a protein with 530 amino acids, is encoded by a gene localized to chromosome 14,q23.2, in close proximity to genes related to Alzheimer’s disease.63,64 Multiple isoforms exist of ER-β, including five full-length forms.
 |
Orphan receptors have been identified that are related to the estrogen receptors. They have been designated as estrogen-related receptor (ERRα, ERRβ, and ERRγ). ERRa may be regulated by coactivator proteins and interacts with typical steroid signaling pathways.65,66 These orphan receptors are expressed in most tissues and may be involved in typical estrogen activities, such as the proliferation and differentiation of target cells in bone and in the breast. Nevertheless, they do not bind estrogens, and no endogenous ligand has been yet identified.
The story is further complicated with the recognition that members of the receptor superfamily are each associated with multiple isoforms.67 This increases the number of possible signaling pathways in physiology and disease. In this discussion, we will mention only the most biologically important isoforms.
The estrogen receptors are divided into six regions in five domains, labeled A to F. The ER-β is 96% homologous in amino acid sequence with the alpha estrogen receptor in the DNA binding domain and 60% homologous in the hormone-binding domain. The full comparison is as follows:63,68,69
ER-α and ER-β Homology (%) | |
The regulatory domain | 18 |
The DNA-binding domain | 96 |
The hinge | 30 |
The hormone-binding domain | 55 |
The F region | 18 |
The hormone-binding characteristics of the ER-α and the ER-β are similar, indicating that they respond in a comparable manner to the same hormones.™ Thus, both receptors bind to the estrogen response element with a similar affinity, and the affinity of estradiol for each receptor is similar. There are differences, however; for example, phytoestrogens have a greater affinity for ER-β than for ER-α. In other words, estrogenic agents demonstrate preferential binding for one or the other receptor. Different genetic messages can result not only because of differences in binding affinity, but also through variations in the mechanisms to be discussed, notably differences in conformational shape and cellular contexts. In addition, because the regulatory domains differ in the two receptors, the ability of ER-β to activate gene transcription by means of TAF-1 is impaired (discussed below).
A/B Region, The Regulatory Domain
The amino acid terminal is the most variable in the superfamily of receptors, ranging in size from 20 amino acids in the vitamin D receptor, to 600 amino acids in the mineralocorticoid receptor. In the ER-a, it contains several phosphorylation sites and the transcription activation function called TAF-1. TAF-1 can stimulate transcription in the absence of hormone binding. The regulatory domain is considerably different in the two estrogen receptors; in ER-β, TAF-1 is either significantly modified or absent.
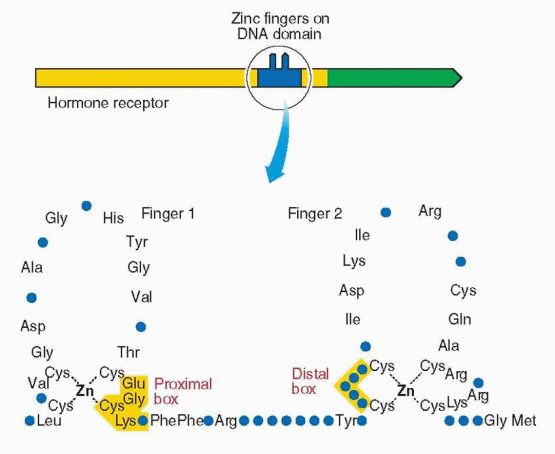
C Region, The DNA-Binding Domain
The middle domain binds to DNA and consists of 100 amino acids with nine cysteines in fixed positions, the two zinc fingers. This domain is essential for activation of transcription. Hormone binding induces a conformational change in the three helices that allows binding to the hormone-responsive elements in the target gene. This domain is very similar for each member of the steroid and thyroid receptor superfamily; however, the genetic message is specific for the hormone that binds to the hormone-binding domain. The DNA-binding domain controls which gene will be regulated by the receptor and is responsible for target gene specificity and high-affinity DNA binding. The specificity of receptor binding to its hormone responsive element is determined by the zinc finger region, especially the first finger. The specific message can be changed by changing the amino acids in the base of the fingers. Substitutions of amino acids in the fingertips lead to loss of function. Functional specificity is localized to the second zinc finger in an area designated the d (distal) box. Different responses are due to the different genetic expression of each target cell (the unique activity of each cell’s genetic constitution allows individual behavior).
 |
D Region, The Hinge
The region between the DNA-binding domain and the hormone-binding domain contains a signal area that is important for the movement of the receptor to the nucleus following synthesis in the cytoplasm. This nuclear localization signal must be present for the estrogen receptor to remain within the nucleus in the absence of hormone. This region is also a site of rotation (hence the hinge designation) in achieving conformational change.
E Region, The Hormone-Binding Domain
The carboxy end of the estrogen receptor-a is the hormone-binding domain (for both estrogens and antiestrogens), consisting of 251 amino acids (residues 302-553). It consists of 12 helices with a folding pattern that forms a pocket where the hormones bind. The pocket is about 20% smaller in ER-β. In addition to hormone binding, this region contains the sites for cofactor binding, is responsible for dimerization, and harbors the transcription activation function called TAF-2. This is also the site for binding by heat shock proteins (specifically hsp 90), and it is this binding to the heat shock proteins that prevents dimerization and DNA binding. In contrast to TAF-1 activity, TAF-2 depends on hormone binding for full activity.
F Region
The F region of ER-a is a 42 amino acid C-terminal segment. This region modulates gene transcription by estrogen and antiestrogens, having a role that influences antiestrogen efficacy in suppressing estrogen-stimulated transcription.71 The conformation of the receptor-ligand complex is different with estrogen and antiestrogens, and this conformation is different with and without the F region. The F region is not required for transcriptional response to estrogen; however, it affects the magnitude of ligand-bound receptor activity.
It is speculated that this region affects conformation in such a way that protein interactions are influenced. Thus, it is appropriate that the effects of the F domain vary according to cell type and protein context. The F region affects the activities of both TAF-1 and TAF-2, which is what one would expect if the effect is on conformation.72
It is speculated that this region affects conformation in such a way that protein interactions are influenced. Thus, it is appropriate that the effects of the F domain vary according to cell type and protein context. The F region affects the activities of both TAF-1 and TAF-2, which is what one would expect if the effect is on conformation.72
Estrogen Receptor Mechanism of Action
Ligand-Dependent Nuclear Activity
The steroid family receptors are predominantly in the nucleus even when not bound to a ligand, except for mineralocorticoid and glucocorticoid receptors where nuclear uptake depends on hormone binding. But the estrogen receptor does undergo what is called nucleocytoplasmic shuttling. The estrogen receptor can diffuse out of the nucleus and be rapidly transported back in or undergo metabolism. When this shuttling is impaired, receptors are more rapidly degraded. Agents that inhibit dimerization (e.g., the pure estrogen antagonists) prevent nuclear translocation and thus increase cytoplasmic degradation.
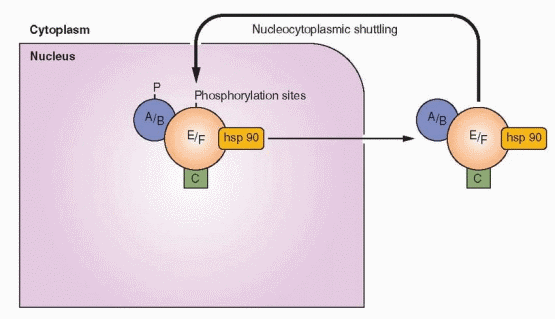 |
In the absence of estrogen (the ligand), the receptor can become associated with the estrogen response element on a gene, a signal for a process that leads to its proteasome degradation via the ubiquitin pathway.73 The receptor bound to its ligand, estrogen, undergoes the same process, but at a pace much slower than unliganded receptor, allowing time for gene transcription. This cyclic turnover allows the target cell to be very sensitive to the concentration of the ligand (estrogen) within the cell.
Prior to binding, the estrogen receptor is an inactive complex that includes a variety of chaperone proteins, including the heat shock proteins. Heat shock protein 90 appears to be a critical protein, and many of the others are associated with it. This heat shock protein is important not only for maintaining an inactive state, but also for causing proper folding for transport across membranes. “Activation” or “transformation” is the dissociation of heat shock protein 90.74
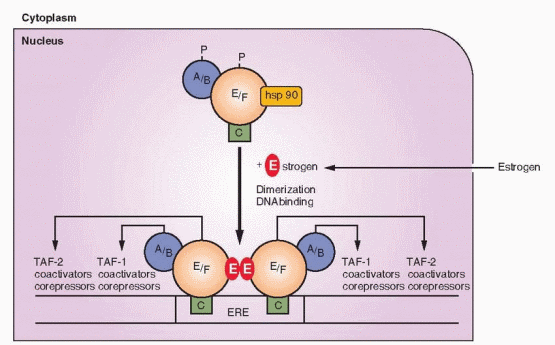 |
Imagine the unoccupied steroid receptor as a loosely packed, mobile protein complexed with heat shock proteins. The steroid family of receptors exists in this complex and cannot bind to DNA until union with a steroid hormone liberates the heat shock proteins and allows dimerization. The conformational change induced by hormone binding involves a dissociating process to form a tighter packing of the receptor. The hormone-binding domain contains helices that form a pocket.75 After binding with a hormone (or with drugs engineered for this purpose), this pocket undergoes a conformational change that creates new surfaces with the potential to interact with coactivator and corepressor proteins. Conformational shape is an important factor in determining the exact message transmitted to the gene. Conformational shape is slightly but significantly different with each ligand; estradiol, tamoxifen, and raloxifene each induce a distinct conformation that contributes to the ultimate message of agonism or antagonism.76,77 Tamoxifen and raloxifene, both TAF-2 antagonists, cause a steric repositioning, about a 90 degree rotation, of a helix (the TAF-2 helix) that then occupies the binding site of a coactivator in those tissues where such a coactivator is a requisite for TAF-2 activity. The weak estrogen activity of estriol is because of its altered conformational shape when combined with the estrogen receptor in comparison with estradiol.78
The hormone-binding domain of the estrogen receptors contains a cavity (the pocket) surrounded by a wedge-shaped structure, and it is the fit into this cavity that is so influential in the final genetic message. The size of this cavity on the estrogen receptor is relatively large, larger than the volume of an estradiol molecule, explaining the acceptance of a large variety of ligands. Thus, estradiol, tamoxifen, and raloxifene each bind in the same cavity within the hormone-binding domain, but the conformational shape with each is not identical.
Conformational shape is a major factor in determining the ability of a ligand and its receptor to interact with coactivators and corepressors. Conformational shapes are not simply either “on” or “off,” but intermediate conformations are possible providing a spectrum of agonist/antagonistic activity. The specific conformational shape of a receptor allows or prevents the recruitment of coactivators and corepressors that ultimately yield various biological reponses.
Members of the thyroid and retinoic acid receptor subfamily do not exist in inactive complexes with heat shock proteins. They can form dimers and bind to response elements in DNA, but without ligand, and they act as repressors of transcription.
Estrogen receptor mutants can be created that are unable to bind estradiol. These mutants can form dimers with natural estrogen receptor (wild type), and then bind to the estrogen response element, but they cannot activate transcription.79 This indicates that transcription is dependent on the result after estradiol binding to the estrogen receptor, an estrogendependent structural change. Dimerization by itself is not sufficient to lead to transcription; neither is binding of the dimer to DNA sufficient.
Molecular modeling and physical energy calculations indicate that binding of estrogen with its receptor is not a simple key and lock mechanism. It involves conversion of the estrogenreceptor complex to a preferred geometry dictated to a major degree by the specific binding site of the receptor. The estrogenic response depends on the final bound conformation and the electronic properties of functional groups that contribute energy. The final transactivation function is dependent on these variables.
Estrogen, progesterone, androgen, and glucocorticoid receptors bind to their response elements as dimers, one molecule of hormone to each of the two units in the dimer. The estrogen receptor-a can form dimers with other alpha receptors (homodimers) or with an estrogen receptor-β (heterodimer). Similarly, the estrogen receptor-β can form homodimers or heterodimers with the alpha receptor. This creates the potential for many pathways for estrogen signaling, alternatives that are further increased by the possibility of utilizing various response elements in target genes. Cells that express only one of the estrogen receptors would respond to the homodimers; cells that express both could respond to a homodimer and a heterodimer.
The similar amino acid sequence of the DNA-binding domains in this family of receptors indicates evolutionary conservation of homologous segments. An important part of the conformational pattern consists of multiple cysteine-repeating units found in two structures, each held in a finger-like shape by a zinc ion, the so-called zinc fingers.80 The zinc fingers on the various hormone receptors are not identical. These fingers of amino acids interact with similar complementary patterns in the DNA. Directed changes (experimental mutations) indicate that conservation of the cysteine residues is necessary for binding activity, as is the utilization of zinc.
The DNA binding domain is specific for an enhancer site (the hormone-responsive element) in the gene promoter, located in the 5′ flanking region. The activity of the hormone-responsive element requires the presence of the hormone-receptor complex. Thus, this region is the part of the gene to which the DNA-binding domain of the receptor binds. There are at least four different hormone-responsive elements, one for glucocorticoids/progesterone/androgen, one for estrogen, one for vitamin D3, and one for thyroid/retinoic acid.81 These sites significantly differ only in the number of intervening nucleotides.
Binding of the hormone-receptor complex to its hormone-responsive element leads to many changes, only one of which is a conformational alteration in the DNA. Although the hormone-responsive elements for glucocorticoids, progesterone, and androgens mediate all of these hormonal responses, there are subtle differences in the binding sites, and there are additional sequences outside of the DNA-binding sites that influence activation by the three different hormones. The cloning of complementary DNAs for steroid receptors has revealed a large number of similar structures of unknown function. It is believed that the protein products of these sequences are involved in the regulation of transcription initiation that occurs at the TATA box.
There are three different RNA polymerases (designated I, II, and III), each dedicated to the transcription of a different set of genes with specific promoters (the site of polymerase initiation of transcription). Transcription factors are polypeptides, complexed with the polymerase enzyme, that modulate transcription either at the promoter site or at a
sequence further upstream on the DNA.82 The steroid hormone receptors, therefore, are transcription factors. The polymerase transcription factor complex can be developed in sequential fashion with recruitment of individual polypeptides, or transcription can result from interaction with a preformed complete complex. The effect can be either positive or negative, activation or repression.
sequence further upstream on the DNA.82 The steroid hormone receptors, therefore, are transcription factors. The polymerase transcription factor complex can be developed in sequential fashion with recruitment of individual polypeptides, or transcription can result from interaction with a preformed complete complex. The effect can be either positive or negative, activation or repression.
In most cases, therefore, the steroid hormone receptor activates transcription in partnership with several groups of polypeptides82:
Other transcription factors—peptides that interact with the polymerase enzyme and DNA.
Coactivators and corepressors—peptides that interact with the TAF areas of the receptor, also called adaptor proteins or coregulators. Previously considered nuclear proteins, these regulators may also have functions within the cytoplasm.
Chromatin factors—structural organizational changes that allow an architecture appropriate for transcription response.
The steroid-receptor complex regulates the amount of mRNA transcripts emanating from target genes. The estrogen-occupied receptor binds to estrogen response elements in the 5′ flanking regions of estrogen-regulated genes, allowing efficient induction of RNA transcription. This can occur by direct binding to DNA and interaction with the estrogen response element or by protein interactions with coactivators between the estrogen receptor and DNA sites. Coactivators and corepressors are intracellular proteins, recruited by hormone receptors, that activate or suppress the TAF areas, by acting enzymatically either on the receptors or on DNA.83,84,85,86,87 and 88 Most of the genes regulated by estrogens respond within 1-2 hours after estrogen administration. Only a few respond within minutes. This time requirement may reflect the necessity to synthesize regulating proteins.89 A large number (over 300—a current list is available at www.nursa.org) of coactivator and corepressor proteins have been identified and designated by code letters and numbers, suggesting that there is a process involved with these proteins, causing selection, activation, queuing, and coordination.90 In general, corepressor proteins bind to hormone receptors in the absence of a ligand and suppress any basal transcription activity. Active investigation of this step will undoubtedly yield understanding of pathologic responses and new pharmacologic developments because it is already recognized that these proteins influence the phenotypes of human diseases.91
Just as coregulators can lead to different responses to the same hormones in different tissues, a new avenue of research indicates that posttranslational modifications of the coregulators can diversify the regulatory effects.88,90 The activity of a coregulator protein can be altered by modifications such as phosphorylation or methylation. The response to a specific hormone, therefore, can be complex, differing in various tissues as directed by the coregulators present in that tissue and how the coregulators are modified. The diversity from tissue to tissue is thus very complex, but even within a single cell impressive diversity is the norm. Because genomic differences among various species are amazingly small (1% or less), evolutionary differences in the way in which genes act, including the complex coregulatory story, make a major contribution to the phenotypic and behavioral differences in life forms, especially humans. In addition, coregulators provide another evolutionary reservoir that can yield adaptation to new environmental challenges and stresses.
The concentration of coactivators/corepressors can affect the cellular response, and this is another explanation for strong responses from small amounts of hormone. With a small amount of receptor but a large amount of coactivator/corepressor, the cell can be very responsive to a weak signal.
One of the aspects of activation, for example with the estrogen receptor, is an increase in affinity for estrogen. This is an action of estrogen, and it is greatest with estradiol and least with estriol. This action of estradiol, the ability of binding at one site to affect another site, is called cooperativity. An increase in affinity is called positive cooperativity. The biologic advantage of positive cooperativity is that this increases the receptor’s ability to respond to small changes in the concentration of the hormone. One of the antiestrogen actions of clomiphene is its property of negative cooperativity, the inhibition of the transition from a low-affinity to a high-affinity state. The relatively long duration of action exhibited by estradiol is due to the high-affinity state achieved by the receptor.
TAF (transcriptional activation function) is the part of the receptor that activates gene transcription after binding to DNA. Ligand binding produces a conformational change that allows TAFs to accomplish their tasks. TAF-1 can stimulate transcription in the absence of hormone when it is fused to DNA; however, it also promotes DNA binding in the intact receptor. TAF-2 is affected by the bound ligand, and the estrogen receptor depends on estrogen binding for full activity. TAF-2 consists of a number of dispersed elements that are brought together after estrogen binding. The activities of TAF-1 and TAF-2 vary according to the promoters in target cells. These areas can act independently or with one another. Indeed, the classic estrogen compounds (e.g., estradiol) produce a conformational shape that allows TAF-1 and TAF-2 to react in a synergistic fashion.
Thus the differential activities of the TAFs account for different activities in different cells. In addition to the binding of the dimerized steroid receptor to the DNA response element, steroid hormone activity is modulated by other pathways (other protein transcription factors and coactivators/corepressors) that influence transcription activation.92,93 This is an important concept: the concept of cellular context. The same hormone can produce different responses in different cells according to the cellular context of protein regulators, and responses can be altered by posttranslational modifications of the cogregulator proteins.
Ligand-Independent Nuclear Activity
Phosphorylation of specific receptor sites is an important method of regulation, as well as phosphorylation of other peptides that influence gene transcription. Phosphorylation can be regulated by cell membrane receptors and ligand binding, thus establishing a method for cell membrane-bound ligands to communicate with steroid receptor genes.
Cyclic AMP and protein kinase A pathways increase transcriptional activity of the estrogen receptor by phosphorylation. In some cases phosphorylation modulates the activity of the receptor; in other cases, the phosphorylation regulates the activity of a specific peptide or coactivator/corepressor that, in turn, modulates the receptor. Phosphorylation follows steroid binding and occurs in both the cytoplasm and nucleus. Thus phosphorylation enhances activity of the steroid receptor complex.
Phosphorylation of the receptor increases the potency of the molecule to regulate transcription. Growth factors can stimulate protein kinase phosphorylation that can produce synergistic activation of genes or even ligand-independent activity. Epidermal growth factor (EGF), IGF-I, and transforming growth factor-alpha (TGF-a) can activate the estrogen receptor in the absence of estrogen, through the TAF-1 domain. This response to growth factors can be blocked by pure antiestrogens (suggesting that a strong antagonist locks the receptor in a conformation that resists ligand-independent pathways). The exact mechanism of growth factor activation is not known, but it is known that a steroid receptor can
be activated by means of a chemical signal (a phosphorylation cascade) originating at the plasma membrane. The recruitment of kinase activity is specific for specific ligands; thus not all ligands stimulate phosphorylation.
be activated by means of a chemical signal (a phosphorylation cascade) originating at the plasma membrane. The recruitment of kinase activity is specific for specific ligands; thus not all ligands stimulate phosphorylation.
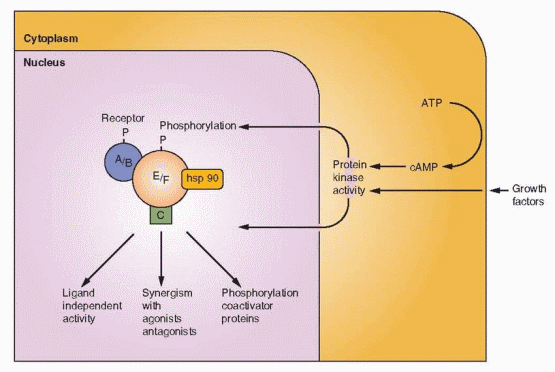 |
Another explanation for strong responses from small amounts of steroids is a positive feedback relationship. Estrogen activates its receptor, gene expression stimulates growth factors (EFG, IGF-I, TGF-a, fibroblast growth factor), and the growth factors in an autocrine fashion further activate the estrogen receptor.94 Ligand-independent activation of the estrogen receptor may be an important mechanism where estrogen levels are low, such as in the male.95
SUMMARY—Steps in the Steroid Hormone-Receptor Mechanism
Binding of the hormone to the hormone-binding domain that has been kept in an inactive state by various heat shock proteins.
Activation of the hormone-receptor complex, by conformational change, follows the dissociation of the heat shock proteins.
Dimerization of the complex.
Binding of the dimer to the hormone-responsive element on DNA at the zinc finger area of the DNA-binding domain.
Stimulation of transcription, mediated by transcription activation functions (TAFs), and influenced by the protein (other transcription factors and coactivators/corepressors) context of the cell, and by phosphorylation.
SUMMARY—Factors that Determine Biologic Activity
Affinity of the hormone for the hormone-binding domain of the receptor.
Target tissue differential expression of the receptor subtypes (e.g., ER-a and ER-β).
The concept of conformational shape: the structure of the ligand-receptor complex, with effects on two important activities: dimerization and the recruitment of regulating proteins.
The concept of cellular context: the differential expression of target tissue regulating proteins, coactivators and corepressors, and phosphorylation, yielding various biological responses.
Different Roles for ER-α and ER-β
Male and female mice have been developed that are homozygous for disruption of the estrogen receptor genes, “estrogen receptor-knockout mice.”68, 96, 97 Estrogen receptor α-deficient mice are known as aERKO or ERKO mice, ER-β deficient mice as αERKO or BERKO mice.98
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


