During pregnancy, the fetus receives via the placenta a continuous source of glucose, which provides energy for metabolic processes and, along with the passage of amino acids, substrate for growth and storage. After delivery, this continuous supply of glucose ceases abruptly and the healthy full-term baby must adapt to intermittent exogenous fuel supply in the fast-feed cycle, while ensuring a continuous internal energy supply for vital organ function.
In this chapter, a summary of the current knowledge of the physiology and pathophysiology of carbohydrate homeostasis and the role of other substrates (carbohydrate as a metabolic substrate must not be considered in isolation) in the neonate is presented. There follows a pragmatic approach to the management of babies with clinically significant hypoglycemia and hyperglycemia and, finally, an overview of the sequelae of poorly controlled glucose homeostasis during a pregnancy complicated by diabetes.
▪ METABOLIC HOMEOSTASIS IN THE HEALTHY MOTHER, FETUS, AND NEONATE
Maternal Metabolism
Maternal metabolic changes during pregnancy are required to support the rapid structural growth of the fetus and its ability to deposit energy stores. In the first trimester, there is increased maternal adiposity, influenced by insulin secretion, which makes available the energy that will be required in the second and third trimesters when daily fetal growth increments and energy requirements increase exponentially. Although maternal insulin secretion increases during the second and third trimesters, the mother becomes relatively insulin resistant as a consequence of the parallel increases in circulating human placental lactogen, progesterone, and estrogen (1). Maternal insulin resistance in turn increases the availability of fuels and substrates for fetal growth and storage, even after short to intermediate periods of maternal fasting.
Fetal Metabolism
During pregnancy, the human fetus receives from its mother, via the placental circulation, a supply of substrates necessary for growth; for the deposition of fuel stores, which are essential after birth (see below); and for energy to meet the basal metabolic rate and requirements for growth. Glucose metabolism accounts for 65% of fetal energy production, with lactate probably accounting for most of the remainder (2). Amino acids also cross the placenta for incorporation into proteins. Fatty acids do not cross the placenta; adipose tissue deposition is via lipogenesis with glucose as the substrate. Studies of perfused human fetal brain have demonstrated that uptake of ketone bodies (the products of betaoxidation of fatty acids) is greater than that of glucose and that the fate of ketone bodies is both incorporation into brain lipids and use as a cerebral energy source (3). However, as evidenced by the adverse neurologic outcomes during poorly controlled diabetes in pregnancy, excessive exposure to a maternal ketotic state may be harmful.
Glucose is transported across the placenta by facilitated diffusion across glucose transporters, whose expression increases as pregnancy develops, in sufficient quantities for metabolic demands and the deposition of glycogen and fat stores, the latter occurring in the third trimester and again regulated by glucose transporters (4). During prolonged maternal starvation or placental insufficiency, the fetus is capable of endogenous glucose production (2). The requirement for fetal glucose production in these circumstances diverts or consumes substrates, first fatty acids and then protein, from growth and storage, and although an immediate protective response, it will ultimately be manifest as fetal growth restriction and associated sequelae. Under extreme circumstances of severe and prolonged placental insufficiency, fetal blood glucose control fails (5). It is possible that such profound and prolonged fetal hypoglycemia has adverse effects on the developing brain and may explain some of the disability following severe intrauterine growth restriction (IUGR), even when there have been no postnatal complications.
In addition to the ability to produce glucose when placental transfer is low (see above), the fetus is able to regulate a high delivery of glucose, for example, if there is suboptimal control of diabetes in pregnancy. In this circumstance, the fetal response to the high placental transfer of glucose is increased secretion of insulin, in turn resulting in greater than normal fetal growth and storage. However, the healthy fetus differs from adults in that there is a blunted insulin response to high glucose concentrations, and insulin secretion is more sensitive to amino acids than glucose, reflecting a greater role of insulin for fetal growth than for fetal glucose control (6). The fetus is less sensitive than is the neonate to the glucose-mobilizing actions of glucagon, although sensitivity increases with gestational age (7,8).
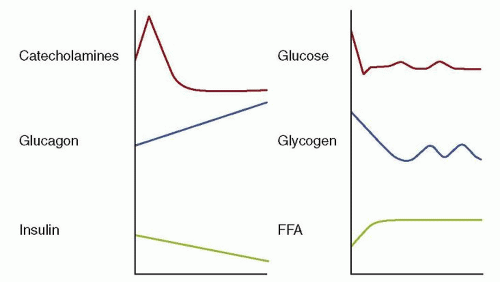
When the continuous flow of nutrients from the placenta is abruptly discontinued, immediate postnatal endocrine and metabolic changes in the healthy baby preserve fuel supplies for vital organ function. Oxygen supply temporarily fails during delivery, so anaerobic metabolism must occur with higher substrate consumption than aerobic metabolism. In addition, the healthy full-term newborn infant must subsequently adapt to the fast-feed cycle and to the change in major energy source, from glucose transferred across the placenta to fat from adipose tissue stores and milk feeds. After birth, plasma insulin levels fall and there are immediate rapid surges of catecholamine and pancreatic glucagon release (7,9). These endocrine changes, in particular the glucagon:insulin ratio, switch on the essential enzymes for glycogenolysis (the release of glucose stored as glycogen in liver, cardiac muscle, and brain), for gluconeogenesis (glucose production from 3-carbon precursor molecules by the liver), lipolysis (release of fatty acids from adipose tissue stores), and ketogenesis (the beta-oxidation of fatty acids by the liver) (10) (Fig. 34.1). Although glucose is the major metabolic fuel for most organs in the hours immediately after birth, there is evidence that lactate may be the preferred cerebral fuel over glucose and ketone bodies at this time (11).
Neonatal Metabolism
The metabolic switch at birth is repeated on a smaller scale during the milk-fed infant’s fast-feed cycles. Immediately after a feed, there is availability of metabolic fuels, namely, fatty acids and, to a lesser extent, sugars from milk. Some tissues, for example, the kidney, are obligate glucose users, but others burn fatty fuels, and the overall respiratory quotient falls after birth, reflecting the fact that fat oxidation accounts for about 75% of oxygen consumption. The neonatal brain takes up and oxidizes ketone bodies at higher rates than seen in adults, and the neonatal brain uses ketone bodies more efficiently than it does glucose (12).
FIGURE 34.1 Levels of hormones and metabolic fuels change after birth. At birth, the counterregulatory hormones (i.e., catecholamines and glucagon) increase greatly, whereas insulin secretion decreases. Neonatal plasma glucose concentrations plummet as a result of cord clamping. The changes in counterregulatory hormones and insulin favor mobilization of glucose and fat and stimulate gluconeogenesis. These changes assure adequate neonatal glucose production. FAA, free fatty acids. From Kalhan SC, Bier DM, Savin SM, et al. Estimation of glucose turnover and 13C recycling in the human newborn by simultaneous [1-13C]glucose and [6,6-1H2]glucose tracers. J Clin Endocrinol Metab 1980;50:456-460, with permission.
Glucose levels peak after a feed; any excess glucose available is then stored as glycogen in the liver or, along with fatty acids absorbed after milk feeds, converted to fat for deposition in adipose tissue. Sometime after each feed, blood glucose level starts to fall and glycogenolysis and gluconeogenesis are again activated to ensure energy availability for organs that are obligate users. Glycogen is an exhaustible source of glucose whose capacity varies according to fetal growth and maturity (13). On average, after approximately 2 hours of fasting, gluconeogenesis must become the major glucose-providing process. Stable isotope turnover studies have shown that neonatal glucose production rates are 4 to 6 mg/kg/min (14). Between feeds, lipolysis and ketogenesis provide alternative fuels to glucose for organs such as the brain, which are not obligate glucose utilizers (15). Indeed, there is evidence that glucose utilization by the neonatal brain is less than in subsequent months, because of utilization of alternative fuels (16). The process of ketogenesis also provides energy and cofactors, which are utilized in gluconeogenesis, again highlighting the importance of fatty fuels.
The control of neonatal metabolism is dependent, first, upon the synthesis of key enzymes, such as hepatic phosphorylase for glycogenolysis, phosphoenolpyruvate carboxykinase (PEPCK) for gluconeogenesis, and carnitine acyltransferases for ketogenesis, and second, upon the induction of enzyme activity by hormonal changes. Glucagon is the major neonatal glucoregulatory hormone (7). The concentration of blood glucagon increases when blood glucose levels fall, and it induces activity of the enzymes of glycogenolysis, gluconeogenesis, and ketogenesis in the liver. The glucoregulatory role of insulin in the neonate is less potent than in the older child and adult (17). In most neonates, insulin does not appear to have a major influence on normal blood glucose homeostasis, but in some extreme cases (see below), high insulin concentrations may result in hypoglycemia. Finally, it is unlikely that other hormones, such as the catecholamines, cortisol, thyroid hormones, and growth hormone, are important regulators in the fastfeed cycle of the healthy neonate, but rare cases of hypopituitarism or cortisol deficiency (see below) may present with neonatal hypoglycemia, which suggests that minimum basal levels are needed to maintain normoglycemia.
Finally, the change from fetal to neonatal metabolism must take into account the important role of gastrointestinal adaptation. The introduction of enteral feeding has been shown to trigger the secretion of gastrointestinal regulatory peptides and hormones, which in turn induce the features of gut adaptation, namely, gut growth, mucosal differentiation, motility, development of digestion and absorption, and even pancreatic hormone responses (18,19).
Differences between Neonatal and Adult Metabolism
The differences between neonatal and adult metabolism are most likely to be evolutionary protective responses. Milk-fed neonates during their normal fast-feed cycle produce and utilize ketone bodies to the extent that is seen in adults only after a prolonged fast. Other fuels such as lactate may also be used in addition to glucose and ketone bodies. Insulin plays a lesser role in neonatal glucoregulation than in the adult, in that its release in response to glucose is blunted and delayed when compared to the adult and that there may be end-organ insensitivities to its action (20). In fact, healthy neonates have insulin-glucose relationships that differ markedly from those of older subjects (21,22).Therefore, when investigating a neonate for possible impaired neonatal glucoregulation, it is essential to have reference data from healthy infants, rather than comparing the neonatal concentrations and interrelationships of fuels and hormones with those of adults. Also, it is inappropriate to consider glucose alone—the availability of alternative fuels must be established.
▪ DISORDERS OF CARBOHYDRATE HOMEOSTASIS
The vast majority of babies are entirely healthy after birth and remain so. For the minority of babies who are born with difficulties or for whom difficulties develop in the hours or days after birth, the etiologies can be described broadly under three headings:
Intrinsic abnormalities acquired during fetal development
Intrauterine, intrapartum, or postnatal external insults that directly injure organs and systems
Failure or delay to make the normal transition from fetal to neonatal life
Taking as an example the cardiovascular system for each of these headings, there may be congenital structural abnormality of the heart acquired during fetal development, there may be myocardial injury and insufficiency arising from a severe hypoxic-ischemic insult, or there may be delayed transition to the normal neonatal circulation with persistent pulmonary hypertension.
With respect to homeostasis of carbohydrate and other metabolic substrates, congenital problems are rare but can cause significant compromise, and severe metabolic disorders arising from external insults are also uncommon. It is the third category (failure or delay to make transition) that is the most common and often significant cause for concern and also for confusion.
Having reviewed, above, the metabolism of the fetus and neonate and the changes that occur at birth in the healthy infant, the three categories of disordered metabolic homeostasis are as follows and in order of increasing occurrence:
Abnormalities acquired during fetal development—for example, inborn errors of metabolism
Intrauterine, intrapartum, or postnatal external insults that directly injure organs and systems—for example, hypoxia-ischemia and infection
Failure or delay in making the normal transition from fetal to neonatal life—for example, following poor control of maternal diabetes mellitus, preterm delivery, or IUGR
Understanding of disordered metabolic homeostasis must be in the context of a knowledge of normal physiology, in order to understand how these disorders will affect a neonate and to assist formulation of sensible management plans.
▪ NEONATAL HYPOGLYCEMIA
Neonatal hypoglycemia has been recognized for over a century (23,24,25), although there have been wide swings of opinion regarding the definition of the condition, its clinical significance, and its optimal management (Fig. 34.2). As described above, healthy babies have a number of protective mechanisms to prevent the physiologic postnatal fall in blood glucose from causing harm. However, some babies have absent, delayed, or impaired protective responses and display clinical signs of hypoglycemia. The risk of reduced glucose availability to the brain in such circumstances has been widely acknowledged (26). However, in more recent years, this risk and the increasing practice of defensive medicine have resulted in a swing towards the treatment of large numbers of infants, often unnecessarily, with intravenous glucose, resulting in separation from their mothers and placing at risk the establishment of breastfeeding (27). Therefore, it is important to identify those infants most at risk for the adverse effects of hypoglycemia and determine the most effective and least invasive regimens for prevention of hypoglycemic brain injury (28). To date, no controlled study has addressed either of these issues.
Diagnosis, Definition and Clinical Significance
Much controversy and confusion has surrounded the definition of hypoglycemia (28,29). Koh et al. (30) demonstrated that the “accepted” definition varied widely not only between standard pediatric textbooks but also between neonatologists, with values given ranging from below 1mmol/L (18mg/dL) to 4mmol/L (72mg/dL). Cornblath et al. (28) wrote, “The definition of clinically significant hypoglycemia remains one of the most confused and contentious issues in contemporary neonatology.” This continuing controversy regarding the definition and clinical significance of neonatal hypoglycemia arises from a frequent failure to consider the changes of metabolic adaptation in their totality (27,28).
The accurate measurement of blood glucose levels is essential in the diagnosis of hypoglycemia. It is well known that glucose reagent strips, commonly used in neonatal and maternity units, are insufficiently reliable for the diagnosis (28). Therefore, if these strips are used for neonatal screening, all low values should be confirmed by accurate measurement. These samples should be assayed promptly as blood glucose levels diminish with time, even in fluoridated tubes (31). Interesting new techniques of glucose monitoring by subcutaneous microdialysis may in time reduce the need for venipuncture and heel pricks (32). However, the clinical significance and validity of glucose measurements using these techniques are not fully evaluated (33).
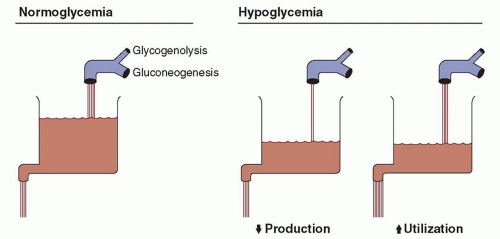
FIGURE 34.2 The rates of glucose production and utilization are represented by the faucet and drain of the sink. The level in the sink is equivalent to plasma or blood glucose concentrations. If production from glycogenolysis and gluconeogenesis is adequate, and use is not excessive, normoglycemia exists and the plasma or blood glucose concentration (i.e., the level in the sink) is normal. Hypoglycemia develops if production is inadequate to meet body needs or if use outstrips production. This results in decreased glucose concentrations (i.e., diminished level in sink). From Kalhan SC, Bier DM, Savin SM, et al. Estimation of glucose turnover and 13C recycling in the human newborn by simultaneous [1-13C]glucose and [6,6-1H2]glucose tracers. J Clin Endocrinol Metab 1980;50:456-460, with permission.
The challenge, in terms of definition, is the description and diagnosis of a pathologic condition, differentiating this from the changes that are within the physiologic “norm” whereby blood glucose levels fall immediately after birth and then increase, often to lower levels than the normal adult range. The increase in blood glucose levels occurs over the first 2 to 3 postnatal days in healthy, appropriate weight for gestational age (AGA) full-term neonates, sometimes later for those who are breastfed. These infants have high ketone body levels when blood glucose concentrations are low, and it is likely that this protects them from neurologic sequelae (15,29,34,35,36). This physiologic pattern in the healthy infant who has no risk factors for impaired metabolic adaptation and has no clinical signs cannot be considered a pathologic condition. Therefore, the definition of the condition we are concerned about is not “a low blood glucose measurement.” A more meaningful definition is “a low blood glucose measurement in the absence of protective metabolic responses.” The level of blood glucose that may be considered “low” is discussed below.
In the absence of accessible and rapid measurements of levels of alternative fuels in the clinical setting, proxy measures for the presence or absence of protective metabolic responses must be considered. In practical terms, this requires identification of risk factors for impaired or delayed metabolic adaptation (Table 34.1) and/or the recognition of abnormal clinical signs that may arise from hypoglycemia, which is not compensated for by alternative fuel utilization (Table 34.2). Therefore, a full and accurate working definition of the condition, which is referred to in the shorthand as neonatal hypoglycemia, should be “a persistently low blood glucose level, measured with an accurate device, in a baby at risk of impaired metabolic adaptation but with no abnormal clinical signs,” or “a single low blood glucose level in any baby presenting with abnormal clinical signs.”
The groups of babies at risk for impaired metabolic adaptation are considered below and in Table 34.1. Signs that may arise from clinically significant hypoglycemia (i.e., a low blood glucose with absent or exhausted metabolic responses) are in Table 34.2. However, no sign is specific to hypoglycemia, and all may arise as the result of coexisting clinical complications, such as perinatal hypoxic-ischemic encephalopathy, or the underlying cause of hypoglycemia (e.g., a metabolic disorder may cause both poor feeding and hypoglycemia).
If early signs of hypoglycemia are not detected and treated, the infant may develop seizures or a reduced level of consciousness. Adverse long-term outcomes have been reported when neurologic signs have been present, although it is difficult to determine the specific impact of hypoglycemia in the presence of preceding or coexisting additional risk factors for brain injury (28,37,38). However, there is evidence from case reports that profound and prolonged hypoglycemia is associated with both transient and permanent structural changes in the brain (39,40,41,42,43,44,45). Gray matter damage is most commonly reported, with the parietooccipital regions being most affected (Figs. 34.3 and 34.4). In extreme cases, profound hypoglycemia, usually the result of serious inborn errors of metabolism, may even result in “cot death” or apparent life-threatening events.
TABLE 34.1 Infants at Risk of Disordered Neonatal Metabolic Adaptation
Preterm (<37 weeks’ gestation)
IUGR—at least 1 of the following:
<2nd centile birth weight
Reduced fat and muscle bulk
Birth weight centile below head circumference centile
Following poor control of maternal diabetes mellitus
Unexplained fetal hyperinsulinism causing clinical appearance of macrosomia
Syndrome, for example, Beckwith-Wiedemann
Moderate to severe perinatal hypoxia-ischemia
Maternal beta-blocker medication
Infection
Known or family history of pituitary or adrenal insufficiency
Known or family history of inborn error of metabolism
No study has yet satisfactorily addressed the duration of absent or reduced availability of metabolic fuels, which is sufficient to cause injury to the brain of the human neonate. Studies in neonatal rats have demonstrated that prolonged insulin-induced hypoglycemia, but not starvation-induced hypoglycemia or a short period of hypoglycemia, resulted in neurodegenerative changes (46). A study of rhesus monkeys has shown that a duration of insulin-induced neonatal hypoglycemia (blood glucose <1.5 mmol/L, 27 mg/dL) of 6.5 hours had no demonstrable long-term effects, whereas 10 hours of hypoglycemia was associated with “motivational and adaptability problems” but no motor or cognitive deficit on testing at 8 months of age (47).
Ideally, an evidence-based definition of hypoglycemia should include the blood glucose concentration considered to be the minimum safe level, the duration beyond which the low blood glucose level is considered to be harmful, the presence of clinical signs, the group of infants studied, the consideration of alternative fuel availability, the conditions of sampling, and the assay methods. Most of these criteria have not been adequately addressed by previous studies or publications and, in reality, will vary between babies (29,37,38). This paucity of data has resulted in a pragmatic approach being proposed by a group of clinicians, that is based on thresholds for intervention rather than attempting to define hypoglycemia as a single numerical term (28). This group proposed that regardless of the blood glucose concentration, neurologic signs in association with low blood glucose levels should prompt investigations to establish a firm diagnosis of hypoglycemia and its underlying cause and the institution of urgent treatment. For infants without clinical signs but at risk of neurologic sequelae by virtue of their impaired ability to mobilize alternative fuels at low blood glucose levels (Table 34.1), intervention to raise blood glucose (measured using an accurate device) should be considered if two consecutive blood glucose levels are below 36 mg/dL (2 mmol/L) or a single blood glucose level is below 18 mg/dL (1 mmol/L).
TABLE 34.2 Signs of Clinically Significant Neonatal Hypoglycemia
Tremor
Irritability
Apnea
Hypotonia
Abnormal cry
Tachypnea
Tachycardia or bradycardia
Pallor
Cyanosis
Hypothermia
Feeding difficulties
Seizures
Excessive drowsiness
Coma
FIGURE 34.3 Term baby, 5 days, admitted at 3 days with poor feeding, clinical seizures, and unrecordable blood glucose. Increased parenchymal signal and loss gray-white matter differentiation in posterior parietal and occipital regions. Courtesy of Dr. N Stoodley.
FIGURE 34.4 Imaging in childhood following neonatal hypoglycemia. Atrophy, gliosis, and ulegyria in posterior parietal and occipital regions.
To summarize the clinical significance of neonatal hypoglycemia, the likely temporal sequence for the baby is as follows, but the glucose thresholds and time periods for clinical signs and potential injury will vary between babies (27):
Low blood glucose levels are found, but the baby does not have clinical signs or sustain injury because the baby is still able to draw on alternative fuel stores, for example, glycogen and fat. This could be defined as biochemical hypoglycemia with adequate metabolic adaptation.
If untreated, the baby exhausts alternative fuel stores and develops subtle clinical signs that are not specific to hypoglycemia (e.g., irritability, lethargy, poor feeding), but hypoglycemia is not damaging at this stage. This is the onset of impaired metabolic adaptation.
If untreated, the baby develops obvious and severe clinical signs (e.g., seizures, coma) but may escape damage if treated very promptly. Metabolic adaptation has failed.
If not treated sufficiently soon after onset of clinical signs, hypoglycemia becomes damaging and in severe cases results in cardiorespiratory arrest.
Finally, the impact of hypoglycemia and its treatment on the mother and baby must be considered. The early neonatal period is an emotionally sensitive time, and the diagnosis of hypoglycemia may create or add to anxiety for the parents. Treatment of the infant with intravenous glucose involves separation of the baby and mother and may be perceived as invasive or painful. The implications for the establishment of breastfeeding must also not be forgotten, especially as there is evidence that separation disrupts breastfeeding and in turn breastfeeding and avoidance of formula supplementation augments ketogenesis (15,35). Therefore, emphasis should be on the early prevention of hypoglycemia and management strategies that do not involve the separation of mother and baby (48).
Mechanisms of Absent, Impaired, or Delayed Metabolic Adaptation in At-risk Groups
Hypoglycemia may be secondary to increased utilization of glucose, to inadequate supply of glucose, or to a combination of the two (Tables 34.3 and 34.4). As described above, this will reach clinical significance if other aspects of metabolic adaptation are also impaired. Depending on the underlying mechanism, the hypoglycemia may be brief and self-limiting requiring supportive treatment only or, more rarely, may be prolonged and require definitive treatment.
TABLE 34.3 Mechanisms of Hypoglycemia
Reduced production
Reduced availability of gluconeogenic precursors
Reduced activity of enzymes of glycogenolysis or gluconeogenesis
Reduced activity of counterregulatory hormones (glucagon, cortisol, catecholamines)
Increased utilization
Hyperinsulinism
Reduced alternative substrate availability
TABLE 34.4 Mechanisms of Hypoglycemia in At-Risk Infants
Reduced production
IUGR
Preterm delivery
Perinatal hypoxia-ischemia
Infection
Inborn errors of metabolism, for example, glycogen storage disorder
Endocrine disorders, for example, hypopituitarism, congenital adrenal hyperplasia/hypoplasia
Maternal beta-blocker medication
Increased utilization
Hyperinsulinism, for example, after poor control of diabetes in pregnancy, Beckwith-Wiedemann syndrome
IUGR—to replenish stores
Reduced availability of alternative substrate, for example, inborn error of fatty acid oxidation
For some babies, there is a clear single cause of hypoglycemia, for example, the baby born after poor control of maternal diabetes who has fetal and neonatal hyperinsulinism. For others, there may be more than one etiologic mechanism (Table 34.4). For example, neonates who have been subject to IUGR may experience impaired glycogenolysis, secondary to low glycogen stores, and impaired gluconeogenesis, secondary to delayed induction of enzymes. Also, their ability to mount a protective alternative fuel response varies in magnitude and sustainability and cannot be relied upon if blood glucose levels are persistently low (49). In addition to factors arising from intrauterine influences, there is evidence that excessive formula milk supplementation may be a cause of the suppressed response, in that ketone body levels are lower in formula-fed infants than in breastfed infants, and there is a negative relationship between ketone body concentration and daily volume of formula taken (35).
It is important to note that not all IUGR infants will be “small for gestational age,” and clinical examination is important for the identification of the “wasted” neonate with disproportionate birth weight and head circumference centiles. Conversely, not all small for gestational age infants will have been subject to placental insufficiency— they may be constitutionally small and will not experience impaired postnatal metabolic adaptation.
Insufficient Supply of Glucose
This is the most common underlying cause of clinically significant neonatal hypoglycemia. In the enterally fed infant, the source of circulating glucose is the absorption and conversion of lactose from milk or, between, feeds from glycogenolysis and gluconeogenesis. Babies receiving intravenous fluids invariably receive glucose as a component of the fluid infusions. If there is insufficient exogenous supply of glucose and the infant fails to switch on glycogenolysis or gluconeogenesis in response to falling blood glucose levels, hypoglycemia will occur. This will be most significant when production of alternative fuels to glucose is also impaired (see above). Three possible mechanisms may cause the failure of glucose production when exogenous supply is too low.
Reduced Availability of Gluconeogenic Precursors
Glycogenolysis and gluconeogenesis may be limited by availability of glycogen, gluconeogenic precursors, or the energy provided by fatty acid oxidation. This may occur after preterm delivery, IUGR, maternal alcohol abuse or perinatal hypoxia-ischemia, or as a consequence of prolonged inadequate intake after birth (50,51,52).
Reduced Activity of Enzymes of Glycogenolysis, Gluconeogenesis, Lipolysis, and Ketogenesis
Despite normal postnatal endocrine responses, there may be failure of synthesis and activation of the key enzymes described above. This may be the result of a specific inherited metabolic disorder, in which case hypoglycemia is usually severe, and recurrent or persistent, or there may be generalized immaturity of enzymes, as in the preterm infant or following IUGR. Finally, enzyme activity may be suppressed by acquired conditions, such as perinatal bacterial infection or impaired liver function secondary to hypoxia-ischemia. Defective gluconeogenesis may also be the cause of hypoglycemia complicating cases of congenital heart disease and cold injury (53,54).
Impaired Counterregulatory Hormone Response
This will result in failure to activate enzymes of glycogenolysis, gluconeogenesis, lipolysis, and ketogenesis. Hyperinsulinism has a dual mechanism in that glucose utilization is increased (see below) but also counterregulatory hormone release is inhibited. Failure of release of counterregulatory hormones (glucagon, catecholamines) may play a role in hypoglycemia in preterm and IUGR babies, and after maternal medication with beta blockers in pregnancy (55,56). Finally, there may be rare permanent disorders that result in insufficiency of counterregulatory hormones, for example, low growth hormone and cortisol levels in septo-optic dysplasia and congenital hypopituitarism and low glucocorticoid levels in adrenocortical deficiencies (57,58,59).
Only gold members can continue reading. Log In or Register to continue