Hirschsprung’s Disease
Jacob C. Langer
University of Toronto, Pediatric General Surgery, Hospital for Sick Children, Toronto, Ontario M5G 1X8, Canada
Hirschsprung’s disease occurs in approximately 1 in 5,000 live-born infants. The disease is characterized by absence of ganglion cells in the myenteric and submucosal plexuses of the intestine, which results in absent peristalsis in the affected bowel. Without normal peristalsis, these children develop a form of functional intestinal obstruction.
The first description of a child with Hirschsprung’s disease appeared in the eighteenth century by Domenico Battini, whose description of a typical child with congenital megacolon was published after Battini’s death in 1800 (1). In 1886, Harald Hirschsprung, a Danish pathologist, described several cases of the condition that ultimately bore his name (2). During subsequent years, surgeons were taught to resect the grossly abnormal colon and perform an anastomosis to the rectum (which rarely worked) or a colostomy. In his textbook on pediatric surgery published in 1926, Fraser said:
This is an obscure disease of the large intestine, in which the essential features are an inability of the colon to part with its contents. … The only curative treatment which the pathology and clinical course of the affection show to be applicable is that of excision of the affected bowel with the union of unaffected gut above to the upper end of the rectum below (3).
Between the turn of the nineteenth century and the 1940s, a number of papers were published that observed abnormalities in the innervation of the colon, but the absence of ganglion cells that we now consider to be the sine quo non of Hirschsprung’s disease was not widely recognized until 1948, when Whitehouse and Kernohan summarized the literature and presented a series of cases of their own (4). Shortly thereafter, Swenson confirmed that aganglionosis was the cause of obstruction in these children and recommended rectosigmoidectomy as the optimal treatment of this disease (5). Although initially this operation was performed without decompressing colostomy in most children (6), technical difficulties in small infants and the debilitated and malnourished state in which many children presented caused most surgeons to adopt a multistaged approach with colostomy as the initial step. In more recent years, advances in surgical technique and earlier diagnosis have resulted in an evolution toward one-stage and minimal access procedures for the treatment of this disease.
ETIOLOGY
Abnormalities in Neural Crest Cell Migration
Early work in the chick embryo by Le Douarin and Teillet suggested that normal ganglion cells originate in the vagal neural crest and migrate from there into the embryonic intestine (7). Subsequent work in several strains of mice that develop congenital aganglionosis suggested there is a delay or arrest in this migration, which results in the neural crest cells failing to reach the distal bowel (8). Later work by other investigators suggested that neural crest cells actually originate in both vagal and sacral sites and migrate toward the middle of the intestine (9,10). This raised the possibility that the neural crest cells get to their destination, but then fail to survive, proliferate, or differentiate due to abnormalities in their microenvironment. Evidence has accumulated of differences in extracellular matrix proteins (11,12), abnormal cell–cell interactions (13), and absence of neurotrophic factors (14) in aganglionic bowel when compared with normal bowel, all of which support the concept that abnormalities in the microenvironment may play a role in producing the distal aganglionosis that is characteristic of Hirschsprung’s disease.
Genetic Abnormalities in Hirschsprung’s Disease
It has long been recognized that Hirschsprung’s disease may have a genetic basis (15). Approximately 10% of children have a positive family history, especially those with longer segment disease. Children with Down syndrome and other genetic abnormalities also have a higher incidence of Hirschsprung’s disease, and the incidence of associated congenital anomalies is approximately 20%. There are a number of clearly defined syndromes that are known to be associated with Hirschsprung’s disease (Table 86-1).
TABLE 86-1 Syndromes and Genetic Abnormalities Commonly Associated with Hirschsprung’s Disease. | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Since the early to mid-1990s, a number of investigators have focused on the genetics of Hirschsprung’s disease (16), and there are now several gene families recognized as being clearly associated with this condition. These include the RET protooncogene, the endothelin family of genes, and several others that are currently under investigation.
The RET protooncogene encodes a tyrosine kinase receptor. Mutations in this gene are known to be involved in the etiology of multiple endocrine neoplasia syndromes type 2, and more recently, different mutations have been associated with some cases of Hirschsprung’s disease. These mutations have been found in 17% to 38% of children with short-segment disease, and in 70% to 80% of those with long-segment involvement (17). Mice in whom the RET protooncogene has been knocked out exhibit absence of renal development and panintestinal aganglionosis (18). There are also a number of other genes that encode for RET ligands, including glial cell line-derived neurotrophic factor (GDNF) and neurturin. Mice in whom either GDNF or neurturin have been knocked out develop ganglion cell abnormalities (19,20), and both GDNF and neurturin mutations have also been found in association with RET abnormalities in a small number of patients with Hirschsprung’s disease (21,22).
The endothelin family of genes was first suspected of being involved in the development of Hirschsprung’s disease during investigation of a large kindred of Mennonites. The endothelin-B receptor and its most important ligand, endothelin-3, are vital to the development of the enteric nervous system, as well as many other neural crest-derived cells. The combination of aganglionosis and piebaldism (caused by melanocyte abnormalities) are present in naturally occurring and endothelin knockout mice (23). This combination, in addition to congenital deafness, is seen in the Waardenburg-Shah syndrome in humans, which has been shown to be due to abnormalities in the endothelin system (24). It is estimated that approximately 3% to 7% of cases of Hirschsprung’s disease are related to this family of genes. Another candidate gene that has more recently been identified is SOX-10, a transcriptional modulator. This gene was found to be mutated in another spontaneous mouse model of aganglionosis, the Dom mouse, and mutations of this gene have subsequently been found in a small number of children with the Waardenburg-Shah syndrome (25).
One additional gene that has been described in a small number of children with Hirschsprung’s disease is SIP1, which encodes for the transcription factor Smad interacting protein 1. These children have a syndrome that includes mental retardation, microcephaly, and distinct facial features (26).
How Do the Genetic Abnormalities Result in Abnormal Neural Crest Cell Migration?
It is unclear exactly how these genetic abnormalities cause abnormal neural crest cell migration and result in the phenotype of Hirschsprung’s disease. It is clear that this is a complex process and that development of the disease is a multigenic phenomenon that can occur at any number of stages during the normal process of neural crest cell migration, differentiation, and survival. There is evidence from animal models that some mutations, particularly those in the endothelin and SOX-10 genes, may produce early maturation or differentiation of neural crest cells, which decreases the number of available progenitor cells and prevents the neural crest cells from migrating further (27,28). There is also evidence that mutations in the RET protooncogene and its related genes likely act by depriving the migrating neural crest cells of an adequately supportive microenvironment (29). Both pathways may also involve apoptosis of migrating neural crest cells (30,31).
CLINICAL PRESENTATION
There are three ways that Hirschsprung’s disease characteristically presents—neonatal bowel obstruction, chronic constipation, and enterocolitis. In addition, some children
have associated anomalies or a recognized syndrome that is associated with an increased risk of Hirschsprung’s disease.
have associated anomalies or a recognized syndrome that is associated with an increased risk of Hirschsprung’s disease.
Neonatal Bowel Obstruction
Approximately 50% to 90% of children with Hirschsprung’s disease present during the neonatal period with abdominal distention and bilious vomiting; there has been a tendency in more recent decades for patients to be recognized earlier. Typically, there is a delay in the passage of meconium; whereas 95% of normal term infants pass meconium in the first 24 hours of life, less than 10% of children with Hirschsprung’s disease have passed meconium during that time. A prenatal history suggestive of intestinal obstruction is rare, except in children with total colonic disease (32). Occasionally, the distal colonic obstruction is so severe that it results in cecal perforation (33). Plain radiographs usually show dilated bowel loops throughout the abdomen. The differential diagnosis of this picture includes all causes of neonatal distal intestinal obstruction, such as jejuno-ileal atresia, meconium ileus or meconium plug syndrome, congenital band, and high anorectal malformation.
Chronic Constipation
Some children are able to manage through the neonatal period and present later with chronic constipation. Commonly, the onset of the constipation is around the time of weaning from breast milk. Although most children who present after the neonatal period have short-segment disease, this history may also be found in those with longer segment or even total colonic involvement, particularly if the child has been exclusively breastfed.
Because constipation is frequently seen in childhood, it may be difficult to differentiate Hirschsprung’s disease from the other, more common causes of constipation. Clinical features that point to this diagnosis include failure to pass meconium in the first 48 hours of life, failure to thrive, gross abdominal distention, and dependence on enemas without significant encopresis. Although many clinicians look for “tightness” of the anal sphincter on rectal examination, this finding is unreliable. Children with functional megacolon often exhibit “stool-holding” behavior and usually date the onset of the constipation to the time of initiation of bowel training.
Enterocolitis
Enterocolitis is characterized by fever, abdominal distention, and diarrhea, and may be life threatening. Approximately 10% of children with Hirschsprung’s disease have enterocolitis as part of the presentation, and because this disease is usually thought of as causing constipation, the diagnosis may therefore be missed. In most cases, the suspicion of Hirschsprung’s disease will be raised if on careful history failure to pass meconium and intermittent obstructive episodes are elicited.
Associated Anomalies and Syndromes
Hirschsprung’s disease may be associated with a wide range of other anomalies, such as malrotation, genitourinary abnormalities, congenital heart disease, limb abnormalities, cleft lip and palate, hearing loss, mental retardation, and dysmorphic features. In addition, it may be part of a large number of recognized syndromes, some of which have an identifiable chromosomal or genetic basis (Table 86-1). Hirschsprung’s disease should therefore be suspected in any child with constipation or neonatal obstruction who is known to have one of these syndromes. In addition, a diagnosis of Hirschsprung’s disease should alert the clinician to the increased possibility of other problems.
DIAGNOSIS
The appropriate diagnostic approach may vary, depending on the age of the patient and the presenting clinical picture. After a careful history and physical examination, the diagnostic steps may include radiographic studies, anorectal manometry, and rectal biopsy.
Radiographic Studies
The first step in the diagnostic pathway for a newborn with a distal bowel obstruction is a water-soluble contrast enema, which can usually rule out intestinal atresia and meconium ileus, the two most common of these diagnoses in addition to Hirschsprung’s disease. In children with Hirschsprung’s disease, the contrast enema may demonstrate a transition zone between the normal and aganglionic bowel (Fig. 86-1). However, because only about 75% of neonates with Hirschsprung’s disease will demonstrate a transition zone (34), the absence of a transition zone does not rule out the diagnosis. In older children with Hirschsprung’s disease, the absence of a transition zone is less common, but may still be present due to a very short aganglionic segment. Other findings on the contrast enema that suggest the diagnosis of Hirschsprung’s disease include a rectosigmoid index (the ratio of rectal diameter/sigmoid diameter) less than 1.0 and retention of barium on a 24-hour postevacuation film.
Anorectal Manometry
Anorectal manometry aids in the diagnosis of Hirschsprung’s disease through identification of the rectoanal inhibitory reflex, which is present in normal individuals but absent in the vast majority of children with Hirschsprung’s disease (Fig. 86-2) (35). Although
anorectal manometry is possible in newborns, it is not widely available for this age group and may be unreliable. In older children, the test is technically easier, but false-positive results may occur due to masking of the relaxation response by contraction of the external sphincter.
anorectal manometry is possible in newborns, it is not widely available for this age group and may be unreliable. In older children, the test is technically easier, but false-positive results may occur due to masking of the relaxation response by contraction of the external sphincter.
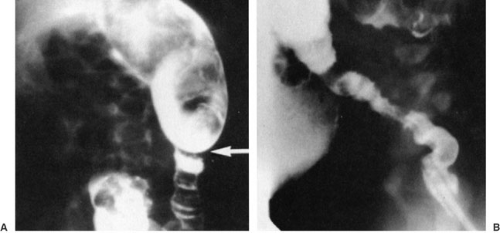 FIGURE 86-1. (A) Contrast enema demonstrating a classic rectosigmoid transition zone in Hirschsprung’s disease. (B) Lateral view of rectum illustrates typical distal spasm of rectim. (Adapted from Sato T, Oldham K. Pediatric abdomen. In: Greenfield LJ. Surgery: scientific principles and practice, 3rd ed. Philadelphia: Lippincott Williams & Wilkins, 2001:2001, with permission.) |
Rectal Biopsy
Definitive diagnosis of Hirschsprung’s disease is based on histologic evaluation of a rectal biopsy, looking for the presence or absence of ganglion cells and the finding of hypertrophied nerve trunks (Fig. 86-3). The biopsy is usually taken 1–2 cm above the dentate line; going too distally may result in a false-positive diagnosis of Hirschsprung’s disease because ganglion cells may normally be absent in this area. The most common technique for rectal biopsy is a suction device that samples mucosa and underlying submucosa. Many pathologists believe that a suction rectal biopsy lacking ganglion cells is consistent with (but is not diagnostic) of Hirschsprung’s disease. Evaluation of suction biopsies may be enhanced by staining for acetylcholinesterase, which has a characteristic staining pattern in the submucosa and mucosa (36). Other stains, such as glial fibrillary acidic protein, have also been described for the diagnosis of Hirschsprung’s disease, but have not been widely adopted (37). Punch biopsies or full-thickness biopsies, which may provide more tissue and deeper levels, may be needed if the suction biopsy sample is inadequate. Some surgeons prefer these techniques as the first choice.
PREOPERATIVE PREPARATION
Once the diagnosis of Hirschsprung’s disease has been established, there are a number of priorities prior to surgical management. Children with dehydration or sepsis must be stabilized and resuscitated using intravenous fluids and antibiotics. Those with intestinal obstruction require a nasogastric tube. Enterocolitis may be relatively mild or may be life threatening. The degree of colonic obstruction can allow for the overgrowth of bacteria, thus leading to diarrhea. If enterocolitis is present, aggressive fluid resuscitation, broad-spectrum antibiotic therapy, nasogastric decompression, and attempts to decompress the obstruction should be instituted quickly.
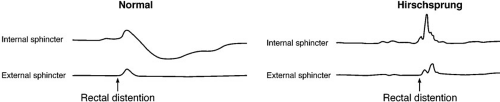 FIGURE 86-2. The anorectal inhibitory reflex. Distension of the rectum with a balloon results in a reflex relaxation of the internal anal sphincter in a normal individual. In a child with Hirschsprung’s disease, there is no reflex relaxation. |
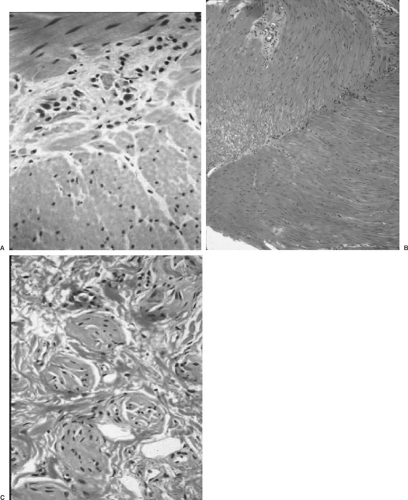 FIGURE 86-3. Histology of Hirschsprung’s disease. (A) Normal ganglion cells in the myenteric plexus. (B) Myenteric plexus without any ganglion cells. (C) Hypertrophied nerve cells that are typical of Hirschsprung’s disease. |
Once a child has been stabilized, surgery can be performed semielectively. While waiting for surgery, most children can be fed breast milk or an elemental formula in combination with rectal stimulations or irrigations. Those that do not tolerate oral or nasogastric feeding can be nourished with parenteral nutrition. In the older child with an extremely dilated colon, weeks or months of irrigations may permit the colon to come down to a more normal size prior to definitive surgery.
SURGICAL OPTIONS
The goals of surgical management for Hirschsprung’s disease are to remove the aganglionic bowel and reconstruct the intestinal tract by bringing the normally innervated bowel down to the anus, while preserving normal sphincter function. There have been many operations devised to accomplish these goals, but the most commonly performed at the present time are the Swenson, Duhamel, and Soave procedures (Fig. 86-4). There are no prospective controlled series comparing surgical treatments of Hirschsprung’s disease. It is therefore difficult to determine if there are any significant advantages to one over the others. It is probably true that surgeons will get the best results doing the operation they have been trained to do and do with some frequency.
The Swenson procedure (Fig. 86-5) is essentially a low anterior resection of the rectum with an end-to-end anastomosis performed by prolapsing the rectum and pulled-through bowel outside the anus. A number of publications have documented excellent results from this approach, including a more recent long-term follow-up of a large group of patients, including some of Swenson’s original patients (38).
Duhamel described a technique in which the native rectum is left in situ and the normally innervated colon is brought behind the rectum in the presacral space. An end-to-side anastomosis is then performed, and the two lumens are joined. Originally, this was accomplished by placing several clamps and cutting between them, but in more recent years most surgeons use a linear stapler for this (Fig. 86-6). The Duhamel procedure has also been widely used around the world, and excellent long-term results have been published (39).
The Soave procedure (or endorectal pull-through) was designed to avoid injury to pelvic vessels and nerves, which are theoretically at risk with the aforementioned procedures, particularly the Swenson procedure. The operation consists of a mucosal proctectomy with preservation of the rectal muscular cuff, and the normally innervated colon is pulled through the muscular cuff and anastomosed just above the dentate line. In the original description, the pulled-through bowel was left hanging out for several weeks, and was then amputated and the anastomosis was completed. Boley’s modification, in which the anastomosis is performed primarily, is the technique usually employed today (40) (Fig. 86-7).
Other approaches that are performed more frequently outside North America include the Rehbein procedure and the use of long myectomy without resection. The Rehbien operation involves a somewhat higher anastomosis than the previously mentioned operations, although long-term follow-up suggests very good results in experienced hands (41). For children with short-segment Hirschsprung’s disease, some surgeons have employed a simple myectomy,
going proximally up to 5 or 6 cm. This can be performed transanally or through a posterior approach. Although good results have been reported in some series, long-term outcomes have not been reported and this approach has not been widely adopted in North America.
going proximally up to 5 or 6 cm. This can be performed transanally or through a posterior approach. Although good results have been reported in some series, long-term outcomes have not been reported and this approach has not been widely adopted in North America.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


