TABLE 43.1 Normal Erythrocyte Values during Gestationa | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
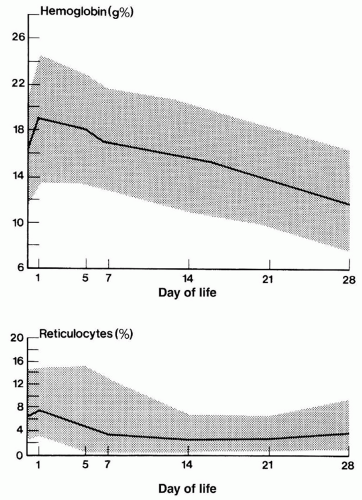 FIGURE 43.1 Hemoglobin values of 178 normal premature infants ≤36 weeks of gestation. Data at the first point, day 0, are cord blood values. Subsequent points represent data from capillary blood samples on 1, 5, 7, 14, and 28 days of life. The dark line represents the mean value, and the shaded area includes 95% of all values. |
1.0 to 1.5 kg, respectively (19,22). Anemia of prematurity largely results from relatively low production of EPO, reduced response to EPO, and blood sampling (23). The signs and symptoms of this early anemia in premature infants are nonspecific and reflect changes in metabolic rate or cardiorespiratory function and perfusion.
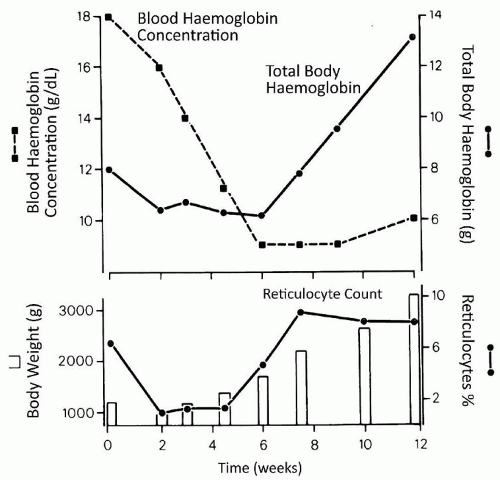 FIGURE 43.2 Changes in total body hemoglobin, blood hemoglobin concentration, reticulocyte count, and body weight in a representative premature infant. The vertical bars represent the infant’s body weight. During the first 6 weeks of life, the blood hemoglobin concentration and total body hemoglobin fall as a result of decreased erythrocyte production, as evidenced by the low reticulocyte count. The more rapid decline in blood hemoglobin concentration from the 3rd to the 6th week is the result of the increasing body size and dilution of the hemoglobin mass. After 6 weeks of age, hemoglobin production increases, as evidenced by the increased reticulocyte count and the rapid increase in total body hemoglobin. The blood hemoglobin concentration during that period may rise slightly, or not at all, because the total body size increases at approximately the same rate as the total hemoglobin mass. |
sampling and an intrauterine transfusion (56). If the hemorrhage has been prolonged or repeated during the course of the pregnancy, anemia develops slowly giving the fetus an opportunity to hemodynamically compensate. These infants may manifest only pallor at birth. After acute hemorrhage just before delivery, the infant may be pale and sluggish, with gasping respirations and signs of circulatory shock.
TABLE 43.2 Types of Hemorrhage in the Neonate | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
possible, and a careful assessment of chorionicity in twins undergoing first trimester ultrasound scanning is recommended (66). The early detection of monochorionic twins identifies a high-risk pregnancy that should be managed in obstetric centers experienced in dealing with such cases. In cases of severe TTTS, the donor (anemic) twin is smaller, and there is associated oligohydramnios; the recipient (polycythemic, hypervolemic) twin is larger, and there is associated polyhydramnios. Intrauterine diagnosis is therefore dependent on identification of same sex, size difference, oligohydramnios/polyhydramnios, and a monochorionic placenta. When diagnosed in utero, TTTS can be classified into five discreet stages, which correlate with probability of survival (67). Stage I is defined by the finding of isolated discrepancy in amniotic fluid volumes between fetuses; absence of a urine-filled bladder in the donor fetus defines stage II, absent or reversed end-diastolic flow in the umbilical artery of the donor fetus or abnormal venous Doppler pattern in the recipient, such as reversed flow in the ductus venosus or pulsatile umbilical venous flow stage III, hydrops fetalis stage IV, and demise of one or both fetuses stage V. Perinatal outcomes correlate with disease severity as assessed by the stage at presentation and gestational age at delivery; overall the perinatal mortality rate for the TTTS is 30% to 50% (68). Therapy has included repeated amniocentesis to reduce polyhydramnios, laser photocoagulation of placental vascular anastomoses, amniotic septostomy, and selective feticide by cord occlusion (66).
monitoring was hidden and represented blood on cotton swabs or in the dead space of syringes or tubing of butterfly sets used to collect blood samples (77).
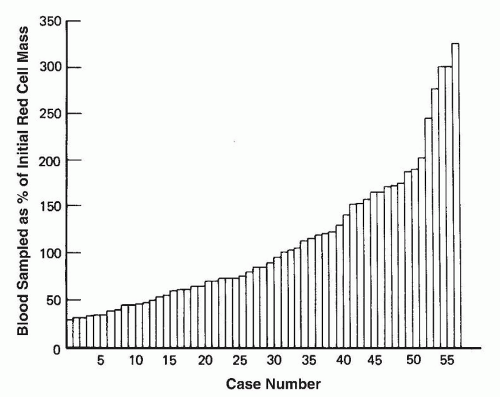 FIGURE 43.3 Cumulative blood losses through sampling in premature infants, expressed as a percentage of their erythrocyte mass at birth. Infants were studied during the first 6 weeks of life, and each vertical bar represents a single infant. |
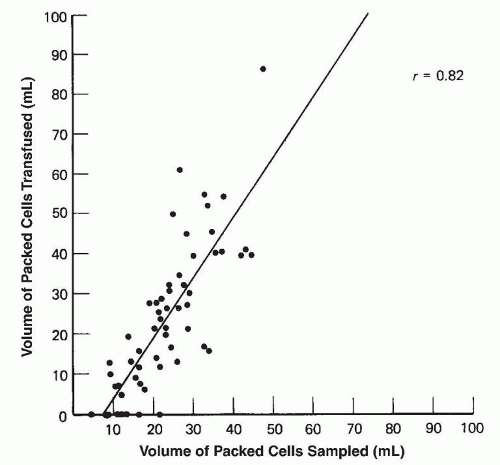 FIGURE 43.4 Relation during the first 6 weeks of life between the cumulative volumes of blood sampled from and transfused into 57 premature infants who had birth weights less than 1,500 g. Volumes represent milliliters of packed erythrocytes (r, correlation coefficient). |
If the infant is pale and limp at birth, clear the airway and assist ventilation.
Obtain venous access immediately usually by insertion of a low umbilical venous line. Blood specimens for complete blood count and crossmatching should be drawn. If an umbilical line is placed, it may be possible to measure a central venous pressure that will be low.
As soon as it is apparent that pallor is a result of hypovolemic shock or profound anemia and not a consequence of asphyxia, administer 15 to 20 mL/kg, depending on ready availability, of O Rh-negative packed RBCs or an isotonic crystalloid solution such as normal saline in the interim. Albumin is no longer recommended for volume replacement (82). Infants with acute external blood loss usually demonstrate dramatic improvement after such a procedure. Infants with massive internal hemorrhages show less evidence of response.
A further infusion of 10 to 20 mL/kg of whole blood or reconstituted whole blood (packed RBCs plus fresh frozen plasma [FFP]) may be given if clinically indicated.
6 months of the delivery of their first child, and another 7.5% show no evidence of immunization 6 months after delivery but develop antibodies during their next pregnancy if their fetus is Rh positive, presumably as a consequence of a sensitization during the first pregnancy.
TABLE 43.3 Some Causes of Hydrops Fetalis | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
prevented by intrauterine transfusions or by the early termination of pregnancy.
characteristic of ABO hemolytic disease. Anemia may be mild or may not be present. Evidence of alloimmunization is difficult to interpret because the DAT may be negative or only weakly positive in up to 40% of cases (111). The diagnosis of ABO hemolytic disease is supported by the finding of increased numbers of spherocytes and increased reticulocyte count. This is in contrast to Rh(D) hemolytic disease of the newborn, which typically presents with anemia, fewer spherocytes, and only minimal increase, if any, in nucleated RBCs (112). The diagnosis of ABO hemolytic disease is supported by the following tests and findings:
Indirect (unconjugated) hyperbilirubinemia
Jaundice appearing during the first 24 hours of life
A group A or B baby of a group O mother
Increased numbers of spherocytes in the blood
Increased erythrocyte production evidenced by reticulocytosis
The presence of IgG, anti-A, or anti-B in cord plasma or serum
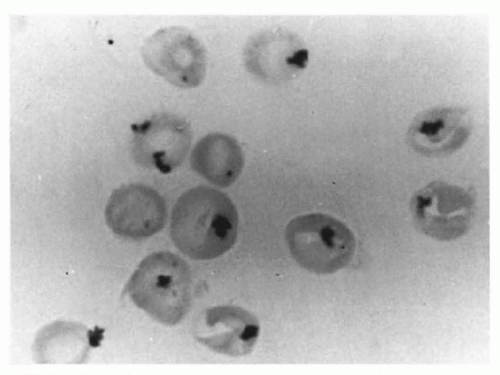 FIGURE 43.5 Heinz bodies in a newborn who developed hemolytic anemia after exposure to naphthalene in mothballs. |
catalase and a relative deficiency of vitamin E. Therefore, newborn infants with G6PD deficiency are at greater risk of developing hemolytic anemia than are adults. G6PD deficiency is associated with an increased incidence of neonatal hyperbilirubinemia, especially if they have class I and class II deficiency. Hyperbilirubinemia in G6PD-deficient newborn males has been reported in Eastern and Western countries (120,121,122,123,124,125,126). It has been reported that male infants of African descent with G6PD deficiency have a significantly higher incidence of hyperbilirubinemia than do controls (127). Although the hyperbilirubinemia is associated with G6PD deficiency, there is a tendency for the jaundice to occur more frequently in particular families and communities, indicating that genetic and environmental factors must influence the incidence of the disease (128).
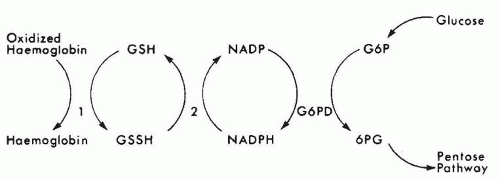 FIGURE 43.6 Protection against oxidative stress in erythrocytes. The erythrocyte is constantly exposed to oxygen; as a result, there is formation of hydrogen peroxide (H2O2), lipid peroxides in the membrane, and oxidized products of hemoglobin such as methemoglobin and Heinz bodies. To prevent the formation of, and to reduce, the levels of these oxidized products, the erythrocyte has a system by which a series of enzyme steps link the metabolism of glucose through the pentose pathway to the reduction of oxidized products (1, glutathione peroxidase; 2, glutathione reductase; G6PD, glucose-6-phosphate dehydrogenase; 6PG, 6-phosphogluconate; G6P, glucose-6-phosphate; GSH, reduced glutathione; GSSH, oxidized glutathione; NADP, nicotinamide adenine dinucleotide phosphate; NADPH, nicotinamide adenine dinucleotide phosphate, reduced). |
TABLE 43.4 Drugs, Chemicals, and Other Factors That Cause Glucose-6-Phosphate Dehydrogenase Deficiency Hemolytic Disease | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
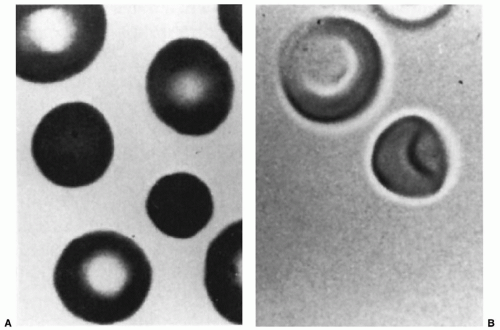 FIGURE 43.7 The erythrocytes of a patient with hereditary spherocytosis, as seen on a stained blood smear (A) and by three-dimensional viewing (B) of glutaraldehyde-fixed cells. |
β-thalassemia) do not manifest during the first month of life. Patients with sickle cell disease are usually found to be anemic by 3 months of age, but cases of jaundice and systemic signs during the neonatal period have been reported (140).
and abnormalities of the thumb including absent or triphalangeal thumbs (155). LBW is seen in about 10% of these patients.
TABLE 43.5 Human Neutrophil Alloantigens (HNA) and Frequencies | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 43.6 Summary of the Inherited Bone Marrow Failure Syndromes and Genes | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
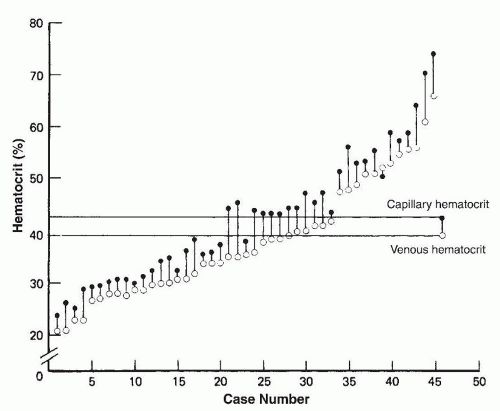 FIGURE 43.8 Simultaneous capillary (dark circles) and venous (open circles) hematocrit levels in 45 premature infants studied during the first 6 weeks of life. Each vertical line represents values for one infant, and the horizontal solid line represents mean capillary and venous hematocrit levels for the whole group. Data are not shown for five infants in whom capillary and venous hematocrit levels were identical. |
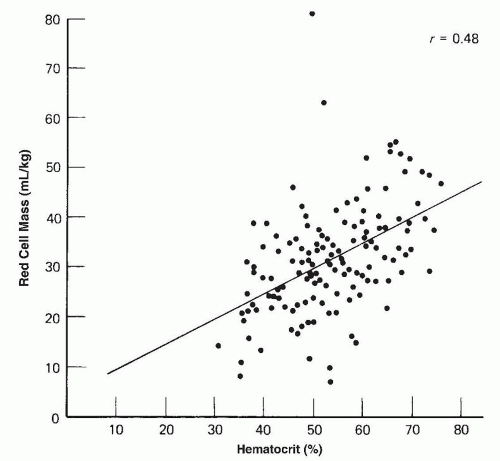 FIGURE 43.9 Simultaneous capillary hematocrit and circulating erythrocyte mass levels in 135 premature infants who had birth weights less than 1,500 g and were studied during the first week of life (r, correlation coefficient). |
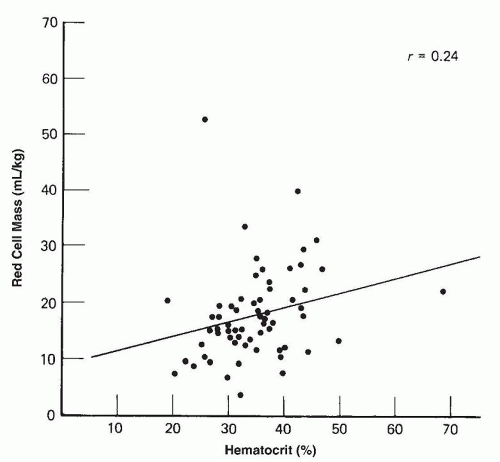 FIGURE 43.10 Simultaneous capillary hematocrit and circulating erythrocyte mass levels in 63 premature infants who had birth weights less than 1,500 g and were studied at 6 weeks of age (r, correlation coefficient). |
the interval between its incision and delivery time >30 seconds as this may have resulted in significant fetal blood loss). Additional questions should be answered. Was the birth traumatic? Did the cord rupture? Was it a multiple birth?
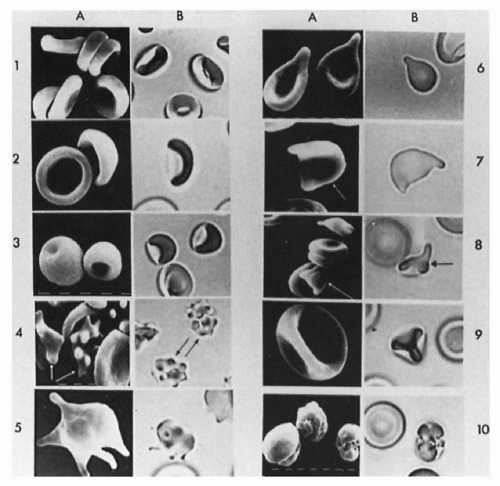 FIGURE 43.11 The three-dimensional appearance of erythrocytes as seen by scanning electron microscopy (A) and by light microscopy (B) of glutaraldehyde-fixed cells (1, discocytes; 2, bowls; 3, spherocytes; 4, echinocytes; 5, acanthocytes; 6, dacrocytes; 7, keratocytes; 8, schizocytes; 9, knizocytes; 10, immature erythrocytes). |
TABLE 43.7 Erythrocyte Differential Counts in Adults and Neonates | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
asphyxia at birth. Polycythemia occurs in 1.5% to 4% of newborn infants (202).
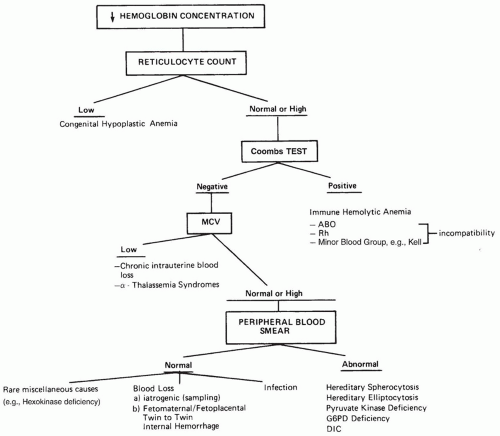 FIGURE 43.12 Diagnostic approach to anemia in the newborn infant. |
is typically drawn from the patient in 10 mL aliquots and replaced with normal saline.
TABLE 43.8 Neonatal Polycythemia | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
an undiagnosed infection, sometimes neutropenia is a sign or a serious underlying disorder that needs urgent diagnosis and treatment.
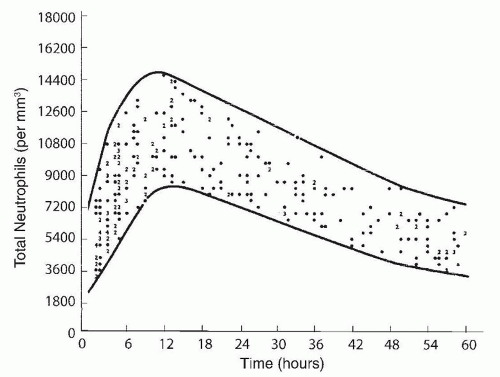 FIGURE 43.13 The total neutrophil count reference range in the first 60 hours of life. Subjects were 434 newborn infants (birth weight 2,685 ± 683 g; range 29-44 weeks of gestational age). Solid circles represent single values; numbers represent the number of values at the same point. Heavy lines represent the envelope bounding these data. Reproduced with permission from Manroe BL, Weinberg AG, Rosenfeld CR, et al. The neonatal blood count in health and disease. I. Reference values for neutrophilic cells. J Pediatr 1979;95:89. |
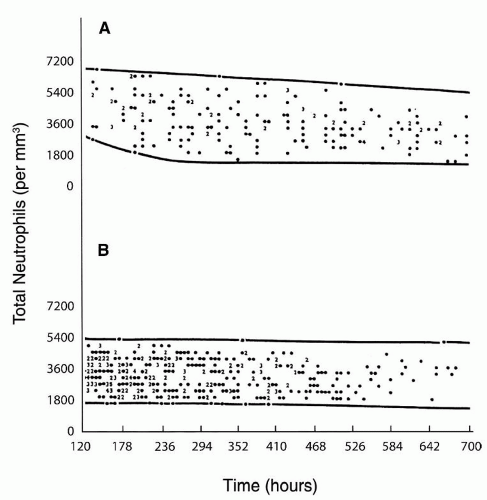 FIGURE 43.14 The reference range for the total neutrophil count for (A) infants 60 to 120 hours of life and (B) 120 hours to 28 days of life. Subjects were 434 newborn infants (birth weight 2,685 ± 683 g; range 29-44 weeks of gestational age). Solid circles represent single values; numbers represent the number of values at the same point. Heavy lines represent the envelope bounding these data. Reproduced with permission from Manroe BL, Weinberg AG, Rosenfeld CR, et al. The neonatal blood count in health and disease. I. Reference values for neutrophilic cells. J Pediatr 1979;95:89. |
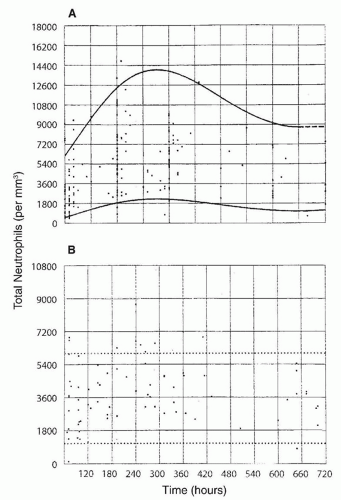 FIGURE 43.15 Reference ranges for total neutrophil values in VLBW infants from (A) birth to 60 hours of life and (B) 61 hours to 28 days of life. Subjects were 193 newborn infants: 50 at 1,000 g and 143 at 1,001-1,500 g. Bold lines (A) and dotted lines (B) represent the envelopes bounding these data, respectively. Reproduced with permission from Mouzinho A, Rosenfeld CR, Sanchez PJ, et al. Revised reference ranges for circulating neutrophils in very-low-birth-weight neonates. Pediatrics 1994;94:76. |
TABLE 43.9 Causes of Neonatal Neutropenia | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
included (215,224), neutropenia was observed in about three-quarters of the subjects. In another study in 65% of 63 neonates with neutropenia and sepsis, the neutropenia was present on the day of the clinical onset of sepsis (224), in 13% of the cases neutropenia developed within 3 days of the onset of sepsis, and in 22% neutropenia was present before the clinical onset of sepsis. Seventy-seven percent of the neutropenic episodes occurred during the first week of life; in 75% of affected neonates, the duration of neutropenia ranged from 0 to 8 days, with 75% having neutropenia for less than 24 hours.
TABLE 43.10 Hematologic Scoring System in Neonates with Suspected Sepsis | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||
bone marrow testing, and genetic testing. Treatment with G-CSF increases neutrophil counts and prevents infection in 90% of cases (255,256). Patients who do not respond to G-CSF may benefit from the addition of low-dose prednisone to the G-CSF regimen or hematopoietic stem cell transplantation (257,258,259). Transformation to myelodysplastic syndrome and acute myeloid leukemia is a major complication in K/SCN regardless of treatment with G-CSF and is an indication for hematopoietic stem cell transplantation.
alloimmune neutropenia was estimated as 1 in 500 newborn infants (288) to less than 0.1% (289). Using modern testing to prospectively analyze 247 cord blood samples of full-term babies and evaluation of neutropenia, the incidence was found to be 0.81% (290).
agglutination test, granulocyte immunofluorescence test, monoclonal antibody immobilization of granulocyte antigens, or a multiplex assay for antibody detection using microbead coupled to purified antigens (292,293,308). These antibody detection assays are best performed in neutrophil reference laboratories and should be complemented by genotyping of the neutrophil-specific antigen of the biologic mother, affected neonate, and/or biologic father acting as a surrogate. It is important to stress that treatment of the infant should not be delayed while confirmatory serologic studies are in progress. Finally, a bone marrow aspirate should be considered if neutropenia is severe (<0.5 × 109/L) and persists for longer than 1 week. Bone marrow biopsy is helpful but may not be feasible in young infants.
TABLE 43.11 Lymphopenia and Immunodeficiency Syndromes in the Neonatal Period | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
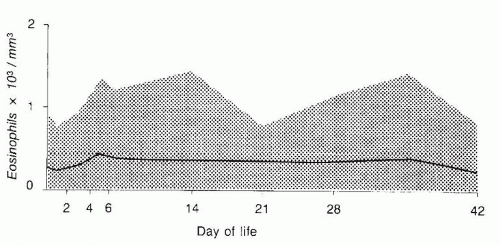 FIGURE 43.16 Eosinophil counts in 142 healthy, premature infants. Data at the first point, day 0, are cord blood values. Subsequent points represent data from capillary blood samples on 1, 5, 7, 14, 28, 35, and 42 days of life. The heavy line represents the mean of each point. The shaded area includes 95% of the infants studied, excluding the top and bottom 2.5% of the group. |
present on the platelets of the mother (autoimmune thrombocytopenia, in which case both the mother and the child may have thrombocytopenia), as occurs in maternal immune thrombocytopenic purpura (ITP) or thrombocytopenia associated with a collagen vascular disorder such as systemic lupus erythematosus.
TABLE 43.12 Causes of Neonatal Thrombocytopenia | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 43.13 Platelet-Specific Alloantigens | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
studies (with a total of 3,028 pregnancies) noted that screening for HPA-1a alloimmunization detects approximately 2 cases of NAIT in 1,000 pregnancies and concluded that screening along with antenatal treatment may indeed reduce the morbidity and mortality associated with NAIT (370). Two additional important points emerge from these prospective screening studies. First, not all pregnancies involving an HPA-1a (PlA1)-alloimmunized mother result in a thrombocytopenic fetus/neonate. Second, HPA-1a (PlA1) alloantibodies may first be detected in the postpartum period as was the case in 39 pregnancies in the large Norwegian study, with the potential to affect future pregnancies (362,363). A recombinant HPA-1a antibody, which competes for binding to the HPA-1a epitope but carries a modified constant region that does not bind to Fcγ receptors, was developed (371). The antibody was studied in healthy HPA-1a1b volunteer subjects and showed improved survival of the subjects’ platelets after ex vivo incubation with the recombinant antibody and reinfusion, as compared to incubation with a destructive IgG1 antibody. The same survival benefit was seen after the platelets were incubated with a mixture of the recombinant antibody and a destructive antibody. The study provides the groundwork for clinical trials studying the ability of the antibody to prevent alloimmunization in HPA-1a-negative mothers.
seem to increase with vaginal delivery (392). Maternal history of ITP, platelet count, and treatment are not necessarily predictive of the newborn’s platelet counts and risk of hemorrhage (393). However, a recent analysis of 127 pregnancies of women with ITP showed that having either a previously affected offspring, or ITP that was refractory to splenectomy are risk factors for significant neonatal autoimmune thrombocytopenia (394). Having a previously affected offspring and a low maternal platelet nadir were found to be significant risk factors in a similar study of 67 neonates (395).
track their platelet nadir and to time appropriate intervention with platelet transfusions. In neonates with NEC, a rapid fall in platelet count to a level well below 100 × 109/L may be a useful marker of intestinal gangrene (422). Finally, in neonates with thrombocytopenia and no apparent cause, thrombosis should be considered.
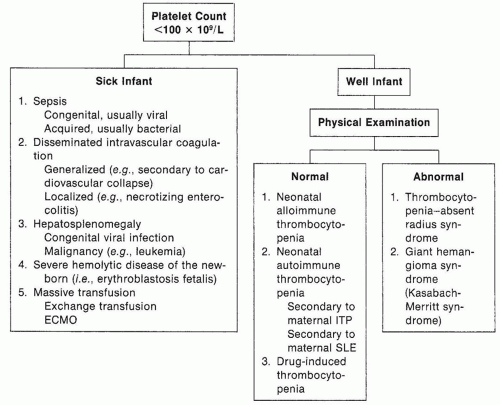 FIGURE 43.17 Approach to the diagnosis of the thrombocytopenic newborn (ECMO, extracorporeal membrane oxygenation; ITP, immune thrombocytopenic purpura; SLE, systemic lupus erythematosus). |
and sequestration in specific organs such as the spleen. In this section of the chapter, the focus is on disorders that have not been covered by other sections.
marrow does not exclude the diagnosis, and genetic testing should be pursued if the clinical presentation is suggestive of the disorder. The cytopenia tends to improve with age (448,449). Using induced pluripotent cells from patients with Pearson syndrome, defects in cell growth and mitochondrial function have been demonstrated during hematopoietic cell differentiation (450).
TABLE 43.14 APTT Reference Values for Neonates and Children Using Four Different Reagents in Two Landmark Studies | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
is reflected in coagulation tests, and the physician should be aware of the expected physiologic differences when interpreting laboratory results (487). Importantly, despite the immaturity, healthy neonates do not tend to develop bleeding or thrombotic complications (488).
TABLE 43.15 TCT, PT, INR, and Fibrinogen Reference Values for Neonates and Children in Two Landmark Studies | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||



