Heart and Lung
Andrew I.M. Campbell
Thomas L. Spray
Department of Cardiothoracic Surgery, Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania 19104.
University of Pennsylvania School of Medicine, Department of Cardiothoracic Surgery, Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania 19104.
Since the mid-1980s, thoracic organ transplantation has become successfully established in the pediatric patient population. Transplantation is now an important option in the treatment of congenital heart and lung disease, as well as the end-stage diseases of these organs. Unfortunately, despite the clinical success in heart and lung transplantation in children, limited donor availability, especially for infant recipients, has prevented more widespread application of this therapy. Pediatric heart and lung transplant recipients make up approximately 10% of all thoracic transplant patients (1,2). In addition, problems such as chronic rejection, graft coronary artery disease (CAD), bronchiolitis obliterans, the infectious and neoplastic complications of current methods of immunosuppression, and the potential limits of transplanted organ growth pose serious questions regarding the long-term utility of thoracic organ transplantation in the pediatric population. This chapter focuses on the clinical aspects of heart, lung, and heart–lung transplantation in infants and children, including indications, preoperative evaluation, operative techniques, postoperative management, and outcome.
HEART TRANSPLANTATION
The first successful neonatal cardiac transplantation was performed by Bailey at Loma Linda (3). This was preceded by his unsuccessful attempt at cardiac xenotransplantation in a newborn. Since 1985, more than 4,500 heart transplantations have been performed worldwide on patients younger than 18 years of age, and 287 were performed in the United States in 2002 (1). There is a bimodal age distribution among heart transplant recipients. A large group of patients are infants with complex congenital heart disease, most of these younger than 2 months of age, whereas a second group is scattered about the early to mid-teenage years as a result of the impact of cardiomyopathy on that age group.
The primary indication for heart transplantation in infancy is complex congenital heart disease without a reasonable corrective or palliative surgical option. The most common congenital anomaly treated by neonatal heart transplantation is hypoplastic left heart syndrome (HLHS), a group of defects characterized by aortic or mitral atresia/stenosis with a diminutive left ventricle. Initial poor results with a staged palliative approach to HLHS led some centers to consider orthotopic heart transplantation as the primary treatment for this anomaly. Long transplant waiting lists have led other institutions to advocate performing a stage I palliation (Norwood procedure) to help stabilize the patient and discontinue the prostaglandins, then to list the patient for heart transplantation (4). However, with improvement in early survival from the Norwood procedure, the majority of cardiac centers have abandoned primary transplantation as initial therapy for HLHS; instead, this option is now reserved for patients in unusually high risk groups, including aortic atresia with a diminutive ascending aorta, severely impaired ventricular function, and severe tricuspid regurgitation (5). Other forms of congenital heart disease that have been treated by cardiac transplantation during infancy include unbalanced atrioventricular canal, single ventricle, complex truncus arteriosus, double-outlet right ventricle, Ebstein anomaly, L-transposition of the great arteries, and pulmonary atresia with intact ventricular septum (6). Primary congenital cardiomyopathy and secondary cardiomyopathies related to rhythm disturbances are also uncommon indications for heart transplantation in infancy.
ABO-incompatible transplantation has been put forward as a method to decrease recipient waiting times and associated waiting list mortality, while increasing the total pool of donors available. In the pediatric cardiac
population, the transplantation of ABO-incompatible organs may be particularly useful because neonates do not yet produce antibodies to T-cell-independent antigens, including the major blood-group antigens. However, with a much greater recipient requirement than available donor pool, application of this process might simply mean that shorter waiting times for some would translate into much longer waiting times for others. In countries where a minority of the infant cardiac donor pool is used, ABO-incompatible transplantation could have a significant effect on minimizing waiting list-related deaths (7).
population, the transplantation of ABO-incompatible organs may be particularly useful because neonates do not yet produce antibodies to T-cell-independent antigens, including the major blood-group antigens. However, with a much greater recipient requirement than available donor pool, application of this process might simply mean that shorter waiting times for some would translate into much longer waiting times for others. In countries where a minority of the infant cardiac donor pool is used, ABO-incompatible transplantation could have a significant effect on minimizing waiting list-related deaths (7).
Children with congenital heart disease who have undergone previous corrective or palliative procedures may exhibit residual or progressive cardiac dysfunction that ultimately requires cardiac transplantation. This group makes up approximately 30% of pediatric heart transplantations from ages 1 to 18 years (1). Cardiac dysfunction is often related to atrioventricular or semilunar valvar insufficiency, which then results in a dilated cardiomyopathy. Even the most complex forms of congenital heart disease, such as heterotaxy syndromes and other anomalies of systemic and venous drainage, are amenable to cardiac transplantation with suitable reconstruction (8). Multiple previous palliative procedures, including those involving the pulmonary arteries, do not preclude successful transplantation (9).
Most pediatric heart transplantations outside infancy are for cardiomyopathy, more than one-half of which are idiopathic in nature. Other etiologies of cardiomyopathy include viral, familial, and hypertrophic. Other less common indications for cardiac transplantation are doxorubicin-induced cardiotoxicity from chemotherapy for malignancy, and obstructive cardiac tumors such as fibromas and rhabdomyomas that are not amenable to surgical resection.
Preoperative Evaluation
The pretransplantation evaluation is a multidisciplinary screening process that is a vital aspect of successful organ transplantation programs. Potential recipients undergo a thorough physical and psychosocial evaluation. The presence of an adequate family support system is of paramount importance. Parents must demonstrate the ability and resources to comply with the complex medical regimens transplant recipients require, and cope with the potential for long or frequent hospitalizations. In addition to this multidisciplinary evaluation, patients undergo screening laboratory tests, including a viral serology panel (e.g., human immunodeficiency virus, cytomegalovirus (CMV), Epstein-Barr virus, hepatitis). The cardiac evaluation is performed by echocardiography and cardiac catheterization. The anatomy of systemic and pulmonary venous connections of the heart and the pulmonary arteries is precisely identified. Important hemodynamic data obtained at catheterization include the systemic cardiac output (Qs) and pulmonary vascular resistance (PVRi), both indexed to the patient’s body area:

Patients with elevated PVRi (greater than 4 to 6 Wood units) are tested with pulmonary vasodilators, including sodium nitroprusside, oxygen (FIO2 100%), and inhaled nitric oxide to establish whether the pulmonary vascular bed is reactive. In general, the presence of a fixed PVRi in excess of 6 to 8 Wood units is a contraindication to orthotopic heart transplantation. Patients who demonstrate improvement with vasodilators may be transplanted with a survival rate comparable to that in patients with normal resistance (9). Although patients with fixed pulmonary hypertension have been successfully transplanted, they have a much higher mortality rate, usually because of postoperative right ventricular failure. Other contraindications to cardiac transplantation include multiple noncardiac congenital anomalies, active malignancy, infection, severe metabolic disease (i.e., diabetes mellitus), multiple organ failure, and the lack of an adequate family support system.
Children with cardiomyopathy are referred for cardiac transplantation for chronic congestive heart failure that limits activity or arrhythmias that are difficult to control. The mortality for idiopathic-dilated cardiomyopathy in children is highest in the first year after diagnosis. Patients with left ventricular shortening fractions of less than 15% by echocardiography and no improvement on follow-up echocardiography have the lowest survival rates, and should be referred for transplantation early (10). The timing for transplantation in children with hypertrophic cardiomyopathy is less clear, because some patients may improve with medication. Those patients with poor diastolic function, despite medical therapy, or recurrent arrhythmias, should be recommended for cardiac transplantation.
Children listed for heart transplantation should be closely monitored until their transplantation, either as outpatients if their condition permits, or while hospitalized. Good nutritional status should be maintained and supplementation such as tube feedings used as needed. A close watch for infectious complications is important, and any subtle indications of infection should be thoroughly investigated. Major infections require patients to have their transplantation status put on hold until they are treated adequately. Anticongestive therapy should be optimized, using digoxin, diuretics, and afterload reduction with captopril or other angiotensin-converting enzyme inhibitors. If heart failure worsens, hospitalization may be required for
inotropic support with dobutamine or phosphodiesterase inhibitors such as milrinone. Long-term therapy may require the placement of an intravenous access device such as a Broviac catheter. The use of extracorporeal membrane oxygenation (ECMO) as a bridge to cardiac transplantation in critically ill children has been limited, mostly to children with postcardiotomy ventricular failure. Although survival rates of up to 50% have been reported, in general, results have been poor (11,12).
inotropic support with dobutamine or phosphodiesterase inhibitors such as milrinone. Long-term therapy may require the placement of an intravenous access device such as a Broviac catheter. The use of extracorporeal membrane oxygenation (ECMO) as a bridge to cardiac transplantation in critically ill children has been limited, mostly to children with postcardiotomy ventricular failure. Although survival rates of up to 50% have been reported, in general, results have been poor (11,12).
The infant or neonate referred for cardiac transplantation requires several other considerations. Infants with complex congenital heart disease such as HLHS are commonly confined to a neonatal intensive care unit (ICU), and are usually maintained on a continuous infusion of prostaglandin E1 to prevent closure of the ductus arteriosus if there is duct-dependent physiology. Balloon atrial septostomy may be helpful if there is a restrictive patent foramen ovale to improve mixing of saturated and desaturated blood, and to decompress the left atrium. Other important issues are the maintenance of adequate nutritional support, avoidance of renal and metabolic complications, and the prompt and thorough treatment of any infectious complications, especially line sepsis, in these fragile infants. Common neonatal problem such as seizures, necrotizing enterocolitis, and intraventricular hemorrhage are also seen in these patients. At the minimum, 10% to 20% of infants die while awaiting a donor heart (8). As mentioned earlier, initial palliative procedures such as the Norwood procedure for HLHS, or Blalock-Taussig shunt for lesions with ductal-dependent pulmonary blood flow, can be performed in the face of a prolonged wait for a donor.
In 2002, the United Network for Organ Sharing revised their classification for pediatric patients awaiting heart transplantation. Status IA applies to patients requiring ventilatory or mechanical circulatory support (i.e., left ventricular assist device (LVAD), extra corporcal membrane oxygenation (ECMO), or balloon pump), multiple- or high-dose inotropes, infants younger than 6 months of age with pulmonary pressures greater than 50% of systemic levels, or any patient with a life expectancy of less than 14 days without a heart transplant. Status 1B applies to patients requiring single-drug inotropic support or infants less than 6 months of age who have significant failure to thrive (less than the fifth percentile for weight and/or height, or loss of 1.5 standard deviations of expected growth). All other patients with less acuity are classified as status 2. A patient’s status may change depending on changes in their clinical condition, or the patient may be placed on hold (status 7) because of infection or other complication, then later reactivated.
Donor Organ Procurement
Criteria for an ideal donor organ are as follows: meets requirement for brain death, consent from next of kin, ABO compatibility, weight compatibility, normal echocardiogram, age younger than 35 years, and normal heart by visual inspection at time of harvest.
The shortage of suitable organ donors, especially for neonatal recipients, has led to many attempts at expanding the donor pool. Hearts from donors with moderately impaired ventricular function by echocardiogram (left ventricular shortening fraction greater than 25%, without major wall motion abnormalities) have been successfully transplanted in infant recipients (8). Donor-to-recipient weight ratios of up to 4:1 have been used in infants, and ischemic times have been successfully extended beyond 9 hours. Deviations from the “ideal” donor criteria should be individualized, and although the use of a marginal donor for a dying infant on ECMO may be justified, the use of the same heart for a child stable as an outpatient would not.
Good donor management is a vital part of successful organ transplantation. The main goals are maintenance of normothermia, euvolemia, adequate tissue perfusion, and prevention of infection. Often, donors with poor cardiac function on initial evaluation will respond to volume loading and low-dose inotropic support with a significant improvement in function following heart retrieval, usually as part of a multiorgan retrieval procedure. The heart is placed at 4°C on ice. In general, cold ischemia time should be limited to a maximum of 4 to 5 hours. The donor heart is placed in cold (4°C), sterile saline, and then triple-bagged in a sterile manner for transport.
Recipient Procedure
Once adequate hemodynamic monitoring is in place and the recipient is properly anesthetized, a median sternotomy is performed and the heart is suspended in a pericardial cradle. If there have been previous sternotomies, appropriate precautions should be taken, including exposing the groins in the sterile field for access for femoral bypass, and the use of an oscillating sternal saw. Once in the chest, the main pulmonary artery is dissected off the aorta past the bifurcation, and the pericardial reflection is mobilized off the aortic arch. In the case of a recipient with HLHS, the aortic arch vessels are mobilized proximally and controlled with snares, and the descending thoracic aorta is dissected to a level 2 to 3 cm below the insertion of the ductus arteriosus. The right and left pulmonary arteries are mobilized and controlled with snares in preparation for cardiopulmonary bypass. After heparinization, the main pulmonary artery is cannulated for arterial inflow, and a single venous cannula is placed in the right atrium because circulatory arrest will be used (Fig. 49-1A). Immediately on instituting cardiopulmonary bypass, the pulmonary arteries are snared tight, perfusing the body through a patent ductus arteriosus. The recipient is cooled to 18°C for circulatory arrest. Once the donor organ is available in the operating room and the patient has been adequately cooled,
circulatory arrest is established, the arch vessels are snared tightly, and the patient is exsanguinated into the venous reservoir. The aorta is divided just above the valve and incised longitudinally along the lesser curve of the aortic arch to a level 1 to 2 cm below the ductal insertion site on the descending aorta (Fig. 49-1B). The ductus is ligated next to the pulmonary artery and divided, then the main pulmonary artery is transected just below the bifurcation. The right atrial incision is started superiorly at the base of the appendage. This incision is then carried down into the coronary sinus and across the atrial septum into the left atrium (Fig. 49-1C). The superior aspect of the right atrial incision is then carried across the septum, opening the roof of the left atrium. The lateral wall of the left atrium is incised above the left pulmonary veins, including the left atrial appendage with the specimen (Fig. 49-1D).
circulatory arrest is established, the arch vessels are snared tightly, and the patient is exsanguinated into the venous reservoir. The aorta is divided just above the valve and incised longitudinally along the lesser curve of the aortic arch to a level 1 to 2 cm below the ductal insertion site on the descending aorta (Fig. 49-1B). The ductus is ligated next to the pulmonary artery and divided, then the main pulmonary artery is transected just below the bifurcation. The right atrial incision is started superiorly at the base of the appendage. This incision is then carried down into the coronary sinus and across the atrial septum into the left atrium (Fig. 49-1C). The superior aspect of the right atrial incision is then carried across the septum, opening the roof of the left atrium. The lateral wall of the left atrium is incised above the left pulmonary veins, including the left atrial appendage with the specimen (Fig. 49-1D).
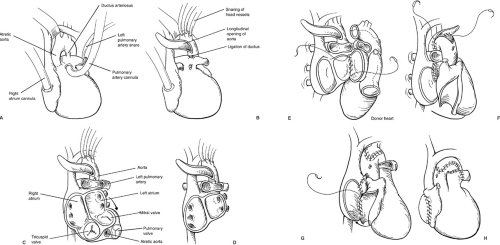 FIGURE 49-1. Recipient procedure. (A) Cannulation of the recipient with hypoplastic left heart syndrome. (B) Longitudinal incision in the recipient aortic arch. (C) Excision of the recipient heart, preserving right and left atrial cuffs. (D) Explantation of recipient heart completed. (E) Implantation of the donor organ beginning at the left atrial anastomosis. (F) Aortic arch reconstruction for the recipient with hypoplastic left heart syndrome. (G) Completion of the right atrial anastomosis. (H) Implantation complete, with decannulation after weaning from cardiopulmonary bypass. |
The donor organ is prepared on the back table in a cold saline solution. The right atrium is incised from the
inferior vena cava laterally to the base of the appendage, avoiding the area of the sinoatrial node. The pulmonary vein confluence is excised off the back of the left atrium, leaving an opening comparable in size to the recipient left atrial cuff. The pulmonary artery is transected just below the bifurcation to provide a wide anastomosis. The aorta is trimmed, depending on the level required in the recipient. Care must be taken to check for and adequately close a patent foramen ovale, which is frequently present, especially in infant hearts. Failure to do so may result in significant postoperative right-to-left shunting in the face of pulmonary hypertension.
inferior vena cava laterally to the base of the appendage, avoiding the area of the sinoatrial node. The pulmonary vein confluence is excised off the back of the left atrium, leaving an opening comparable in size to the recipient left atrial cuff. The pulmonary artery is transected just below the bifurcation to provide a wide anastomosis. The aorta is trimmed, depending on the level required in the recipient. Care must be taken to check for and adequately close a patent foramen ovale, which is frequently present, especially in infant hearts. Failure to do so may result in significant postoperative right-to-left shunting in the face of pulmonary hypertension.
The implantation is begun by anastomosing the lateral wall of the left atrium, from the level of the left atrial appendage inferiorly (Fig. 49-1E). A left ventricular vent is placed through the right superior pulmonary vein, and the left atrial anastomosis is completed by reconstructing the intraatrial septum. The arch of the aorta is then reconstructed (Fig. 49-1F). The right atrial anastomosis is begun at the inferior vena cava orifice, then taken superiorly along the intraatrial septum (Fig. 49-1G). Once the right atrial anastomosis is completed, the ligature is removed from the donor superior vena cava and the venous cannula is placed through the vena caval stump. The
ascending aorta is then cannulated by a new pursestring, air is evacuated, and cardiopulmonary bypass resumed. The snares are released from the head vessels and warming is commenced. The pulmonary anastomosis is then performed in an end-to-end fashion. If time permits, this may be done during circulatory arrest in a drier field. After adequate warming, the patient is weaned from cardiopulmonary bypass and the cannulas removed (Fig. 49-1H). Right atrial, left atrial, and occasionally pulmonary artery pressure catheters are placed before discontinuing bypass, and brought out through the skin below the incision.
ascending aorta is then cannulated by a new pursestring, air is evacuated, and cardiopulmonary bypass resumed. The snares are released from the head vessels and warming is commenced. The pulmonary anastomosis is then performed in an end-to-end fashion. If time permits, this may be done during circulatory arrest in a drier field. After adequate warming, the patient is weaned from cardiopulmonary bypass and the cannulas removed (Fig. 49-1H). Right atrial, left atrial, and occasionally pulmonary artery pressure catheters are placed before discontinuing bypass, and brought out through the skin below the incision.
In older children with cardiomyopathy or infants without aortic arch abnormalities, the recipient procedure is similar to that performed on adults. The ascending aorta is mobilized to the pericardial reflection and is used for arterial cannulation. The child is cooled to 26°C to 28°C because the implantation is done under aortic cross-clamp, rather than circulatory arrest. After the left atrial anastomosis has been completed, the right atrial connection can either be sewn directly or by using a bicaval technique, if a previous cavopulmonary connection has been performed. This may decrease the incidence of tricuspid regurgitation in certain patients. The aortic anastomosis is then completed in an end-to-end fashion in the mid-ascending aorta. The pulmonary artery anastomosis may or may not be performed during aortic cross-clamp, depending on the amount of time the implant takes.
There are numerous other variations of the implantation procedure, depending on the recipient anatomy present. Modifications accounting for a persistent left superior vena cava, prior cavopulmonary shunt or Fontan procedure, corrected transposition of the great arteries, and situs inversus totalis have all been described, but are beyond the scope of this chapter (8,13).
Postoperative Management
The recipient is returned from the operating room to an isolation room in the ICU. Mechanical ventilation is required initially, but is weaned as rapidly as possible. Antibiotics are continued until all monitoring lines and chest tubes have been removed. In older patients, early ambulation is encouraged.
Some level of inotropic support is required in virtually all heart transplant recipients. Isoproterenol is often an ideal choice because of the inotropic and chronotropic effects because many patients have a slower-than-optimal heart rate initially. Dobutamine and dopamine, especially at “renal dose,” are also frequently used. Epinephrine and norepinephrine are usually reserved for poor graft function. Sodium nitroprusside infusion is used for afterload reduction in the early postoperative period. Right ventricular dysfunction due to pulmonary hypertension may respond to phosphodiesterase inhibitors such as milrinone. Inhaled nitric oxide has been shown to be an effective selective pulmonary vasodilator with few systemic side effects and is useful in cardiac transplant recipients with pulmonary hypertension.
TABLE 49-1 Heart Transplantation Immunosuppression Regimen. | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Standard triple-drug (prednisone, cyclosporine, azathioprine) immunosuppression therapy has been successfully used in pediatric cardiac transplant recipients (14). The induction and maintenance doses of medications used for immunosuppression at the Children’s Hospital of Philadelphia are listed in Table 49-1. Because of the adverse effects of corticosteroids, withdrawal from prednisone is usually attempted at 6 months posttransplantation. It has been shown that up to 80% of patients may be successfully weaned from steroids; only one-fourth of these patients have an episode of rejection in the first 6 months (15). An increasing number of centers recommend the use of induction immunosuppression in pediatric cardiac recipients with close to 40% of patients now receiving either a polyclonal anti-T cell preparation, OKT3 (a murine monoclonal CD3 antibody), or an interleukin-2 receptor antibody immediately following transplantation (1).
Tacrolimus (formerly called FK-506) has been shown by the Pittsburgh group (16) to be an effective immunosuppressive agent in children, and its usage has increased over the last 5 years with approximately 40% of all pediatric cardiac transplant patients receiving it for maintenance immunosuppression 1 year after transplantation.
Tacrolimus (formerly called FK-506) has been shown by the Pittsburgh group (16) to be an effective immunosuppressive agent in children, and its usage has increased over the last 5 years with approximately 40% of all pediatric cardiac transplant patients receiving it for maintenance immunosuppression 1 year after transplantation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


