Genetic Lesions in Steroidogenesis
Walter L. Miller
The key clinical, laboratory, and therapeutic features of the common forms of congenital adrenal hyperplasia (CAH) are discussed in this chapter. Manifestations of a deficiency of each enzyme in the pathway, including the clinical presentation, laboratory findings, and therapeutic measures, are shown in Table 533-1. Because each steroidogenic enzyme has multiple activities and many extra-adrenal tissues contain enzymes that have similar activities, the complete elimination of a specific adrenal enzyme may not result in the complete elimination of its steroidal products from the circulation.
LIPOID CONGENITAL ADRENAL HYPERPLASIA
Lipoid congenital adrenal hyperplasia (CAH) is the most severe genetic disorder of steroid hormone synthesis. This disorder is characterized by the absence of significant concentrations of all steroids, high basal corticotropin (ACTH) and plasma renin activity, an absent steroidal response to long-term treatment with high doses of ACTH or human chorionic gonadotropin (hCG), and grossly enlarged adrenals laden with cholesterol and cholesterol esters.1-4
 EPIDEMIOLOGY
EPIDEMIOLOGY
Lipoid congenital adrenal hyperplasia (CAH) is common in Japan and Korea.4-13 The carrier frequency for this mutation appears to be about 1 in 30012 so that 1 in every 250,000 to 300,000 newborns in these countries is affected. Other genetic clusters are found among Palestinian Arabs, in eastern Saudi Arabia,8 and in Switzerland.14 Rare mutations in P450scc and steroidogenic factor 1 (SF1) can produce a very similar clinical picture.
 CLINICAL FEATURES
CLINICAL FEATURES
In most cases of lipoid congenital adrenal hyperplasia (CAH), an infant with normal-appearing female genitalia experiences failure to thrive and salt loss in the first weeks of life,4,11 although some patients have presented later,8 sometimes with apparent sudden infant death syndrome (SIDS).15 A milder form of “nonclassic lipoid CAH” has been described in children with symptoms of adrenal insufficiency at 2 to 4 years of age.16
 PATHOPHYSIOLOGY
PATHOPHYSIOLOGY
Lipoid congenital adrenal hyperplasia (CAH) results from a lesion in the first step in steroidogenesis—the conversion of cholesterol to pregnenolone. However, placental steroidogenesis persists in lipoid CAH, permitting normal term gestation, suggesting that P450scc is not involved.15 Loss of steroidogenic acute regulatory protein (StAR) leads to a loss of most, but not all steroidogenesis, with a compensatory rise in corticotropin (ACTH) and luteinizing hormone (LH). This stimulates uptake of low-density lipoprotein (LDL) cholesterol and de novo synthesis of cholesterol that accumulates in the mitochondria of steroid-producing cells, and as in a storage disease, the cholesterol, cholesterol esters, and their autooxidation products cause cellular damage in the adrenal and fetal testis, as shown in eFigure 533.1  .6 The absence of fetal testosterone production leads to male pseudohermaphroditism.
.6 The absence of fetal testosterone production leads to male pseudohermaphroditism.
 TREATMENT
TREATMENT
Treatment of lipoid congenital adrenal hyperplasia (CAH) is straightforward if the diagnosis is made. Physiologic replacement with glucocorticoids, mineralocorticoids, and salt will permit survival to adulthood.3,4 The glucocorticoid requirement is less than in the virilizing adrenal hyperplasias because it is not necessary to oversuppress excess adrenal androgen production; thus, growth in these patients should be normal.4
3β-HYDROXYSTEROID DEHYDROGENASE DEFICIENCY
3βHSD deficiency is a rare cause of glucocorticoid and mineralocorticoid deficiency that is fatal if not diagnosed early in infancy.20 In its classic form, genetic females with 3bHSD deficiency have clitoromegaly and mild virilization because the fetal adrenal overproduces large amounts of dehydroepiandrosterone (DHEA), a small portion of which is converted to testosterone by extraadrenal 3βHSD1. Genetic males also synthesize some androgens by peripheral conversion of adrenal and testicular DHEA, but the concentrations are insufficient for complete male genital development; thus, these males have a small phallus and severe hypospadias.21-30,31
17α-HYDROXYLASE/17,20-LYASE DEFICIENCY
P450c17 is the single enzyme that catalyzes both 17α-hydroxylase and 17,20-lyase activities. 17α-Hydroxylase deficiency is especially common in Brazil.28,29,31-37 Deficient 17α-hydroxylase activity results in decreased cortisol synthesis, overproduction of corticotropin (ACTH), and stimulation of the steps proximal to P450c17. These patients may have mild symptoms of glucocorticoid deficiency, but this is not life threatening because the lack of P450c17 results in the overproduction of corticosterone, which also has glucocorticoid activity.36 Affected patients also typically overproduce DOC, a mineralocorticoid, in the zona fasciculata, causing sodium retention, hypertension, and hypokalemia, affected females are phenotypically normal but fail to undergo adrenarche and puberty,40 and genetic males have absent or incomplete development of the external genitalia (male pseudohermaphroditism). The classical presentation is that of a teenage female with sexual infantilism and hypertension. The diagnosis is made by finding low or absent 17-hydroxylated C-21 and C-19 plasma steroids.
21-HYDROXYLASE DEFICIENCY
 EPIDEMIOLOGY
EPIDEMIOLOGY
21-Hydroxylase deficiency is one of the most common inborn errors of metabolism, occurring in about 1 in 15,000 births,41-46 and accounts for about 95% of all forms of congenital adrenal hyperplasia (CAH). The overall incidence of “classical” CAH (ie, salt-wasting and simple virilizing CAH) is 1 in 15,000 live births in Caucasians and Hispanics, and 1 in 42,300 African Americans.46 Nonclassical CAH (hirsutism, virilism, menstrual irregularities, and decreased fertility in adult women; so-called late-onset CAH) is reported to have incidences of 1 in 27 for Ashkenazi Jews, 1 in 53 for Hispanics, 1 in 63 for Yugoslavs, 1 in 333 for Italians, and 1 in 1000 for other whites.47-57
 GENETICS
GENETICS
Detailed reviews of the molecular genetics of this disorder are available.47 There are two 21-hydroxylase loci, a functional CYP21A2 gene, generally termed 21B, and a nonfunctional CYP21A1P pseudogene, termed 21A.59 These genes are duplicated in tandem with the C4A and C4B genes encoding the fourth component of serum complement.60,61 Because they lie in the human leukocyte antigen (HLA) locus with its very high rate of genetic recombination, the genetics of 21-hydroxylase deficiency is based on recombination. 21-Hydroxylase deficiency can be caused by 21B gene deletions, gene conversions, and apparent point mutations, which are typically small gene conversion events.62-84 Most patients with 21-hydroxylase deficiency are compound heterozygotes, having different lesions on their two alleles. Because gene deletions and large conversions eliminate all 21B gene transcription, in the homozygous state these lesions will cause salt-losing CAH. Cloning of mutant 21B genes causing CAH shows that a relatively small number of mutations cause CAH, virtually all of which are also found in the 21A pseudogene. These observations indicate that most CAH alleles bearing apparent point mutations actually carry microconversions.84 Most patients are compound heterozygotes, carrying a different mutation on the allele inherited from each parent. Extra-adrenal 21-hydroxylases can influence the clinical phenotype, as can variations in androgen metabolism and sensitivity. Thus, discordances between genotype and phenotype are to be expected.
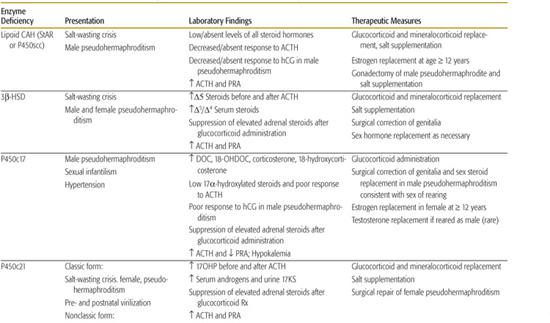
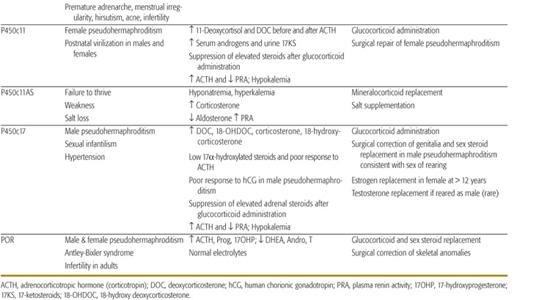
Table 533-1. Clinical and Laboratory Findings and Therapeutic Measures in the Congenital Adrenal Hyperplasias
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


