Disorders of Branched Chain Amino and Organic Acid Metabolism
Stephen I. Goodman and Jean-Marie Saudubray
CATABOLISM OF BRANCHED-CHAIN AMINOACIDS
The three essential branched chain aminoacids (BCAAs), leucine, isoleucine, and valine encompass about 25% of human protein. They are metabolized in mitochondria. The first two catabolic steps are common to the three BCAAs (Fig. 137-1). The first reaction, which occurs primarily in muscle, involves reversible transamination to 2-oxo (or keto) acids and is followed by oxidative decarboxylation to coenzyme A (CoA) derivatives by branched-chain oxo (or keto) acid dehydrogenase (BCKD). BCKD is similar in structure to pyruvate dehydrogenase, is also highly regulated by a kinase/phosphatase system and plays a key role in nitrogen metabolism.
Subsequently, the degradative pathway of BCAAs diverge. Leucine is catabolized to aceto-acetate (Ketogenic amino acid) and acetyl-CoA which enters the Krebs cycle. The final step in the catabolism of isoleucine involves cleavage into acetyl-CoA and propionyl-CoA, which also enters the Krebs cycle via conversion to succinyl-CoA. Valine is also ultimately catabolized to propionyl-CoA.
The most frequent inborn errors affecting the BCAAs catabolic pathway are maple syrup urine disease (MSUD), isovaleric, propionic, and methylmalonic acidemias (or acidurias). These four disorders can present either as a neonatal metabolic crash, as a late onset acute intermittent form, or as a chronic progressive form. Many other rare disorders have also been described on this catabolic pathway. All these disorders can be diagnosed by identifying acylcarnitines and other organic compounds in plasma and urines by gas chromatography-mass spectrometry (GC-MS) or tandem MS and all can be detected by newborn screening using tandem MS.
MAPLE SYRUP URINE DISEASE
In maple syrup urine disease (branched-chain ketoaciduria), major cerebral symptoms appear early in the newborn period, and the urine has an odor reminiscent of maple syrup. The branched-chain amino acids—leucine, isoleucine, and valine—are present in high concentration in the blood and urine, and the ketoacid analogues are found in the urine.
 CLINICAL FINDINGS
CLINICAL FINDINGS
Infants with maple syrup urine disease (MSUD) appear well at birth. In the typical patient, symptoms begin after 3 to 5 days and progress rapidly to death within 2 to 4 weeks.1 Early manifestations include feeding difficulty, irregular respirations, progressive loss of the Moro reflex and lethargy. Severe hypoglycemia may occur. Characteristically these patients develop convulsions, opisthotonos, and generalized muscular rigidity, boxing and pedalling movements with or without intermittent flaccidity. Death usually occurs after decerebrate rigidity develops. Cortical atrophy may be seen on CT or MRI scan, and the myelin is usually hypodense. This is consistent with the defective myelinization that has been observed at autopsy. The feature that distinguishes any form of branched-chain ketoaciduria from other cerebral degenerative diseases of infancy is the characteristic maple syrup, or caramel, odor of the urine, skin, or hair. The odor may become evident 1 or 2 days after birth and may persist, but varies in intensity and may not be detected in some specimens.
Milder forms of the disease occur that are known as intermittent branched-chain aminoaciduria; they represent variant mutations in the same enzyme complex as in classic maple syrup urine disease. Ataxia and repeated episodes of lethargy with ketoacidosis progressing to coma occur without mental retardation; these episodes may be precipitated by infection or anesthesia.
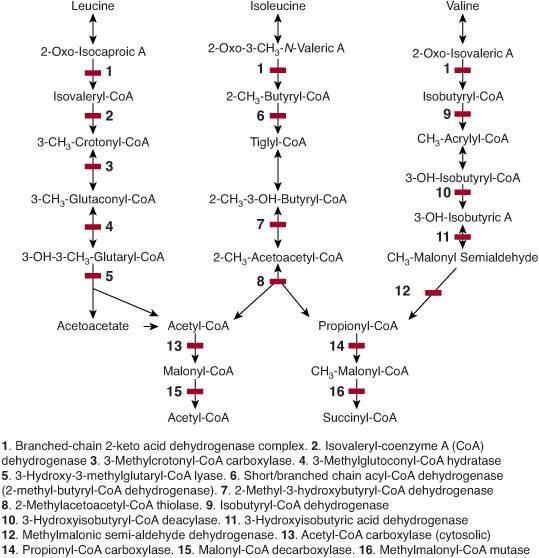
FIGURE 137-1. Metabolism of leucine, isoleucine, and valine. The enzymes involved in the stages of metabolism are numbered.
 BIOCHEMICAL FINDINGS, GENETICS
BIOCHEMICAL FINDINGS, GENETICS
Increased quantities of leucine, isoleucine, and valine are found in the plasma and urine. The presence of an abnormal amino acid, alloisoleucine, is diagnostic for MSUD. The catabolism of branched-chain amino acids is initiated by a transamination reaction to generate the respective ketoacids which then undergo decarboxylation to coenzyme A (CoA) derivatives. The defect in MSUD is in this oxidative decarboxylation of the ketoacids, which is catalyzed by a mitochondrial multienzyme complex similar to pyruvate dehydrogenase and to α-ketoglutarate dehydrogenase. For this reason, autosomal recessive mutations in four different genes can cause MSUD. Patients have been identified with defects in the E1α, E1β, E2, and E3 subunits of the complex. The E3 subunit is shared by all three complexes, and patients with defects in this gene have simultaneous deficiency of branched-chain ketoacid dehydrogenase, pyruvate dehydrogenase, and α-ketoglutarate dehydrogenase. MSUD is rare in most populations with an incidence of ∼1/150,000.
 THERAPY
THERAPY
Experience has now been accumulated with prolonged use of special diets in which the intakes of leucine, isoleucine, and valine are closely controlled. Concentrations of the branched-chain amino acids in plasma can be maintained within normal limits. This therapy is difficult. Many patients have had permanent brain damage before treatment is started, but experience with siblings of previous patients, in whom very early diagnosis is possible, and with patients detected by neonatal screening programs, indicates that a normal IQ may be achieved. Commercial products are available that are useful in management. Intravenous solutions of amino acids that exclude the branched-chain amino acids take advantage of protein synthesis to reduce concentrations of leucine and the other amino acids and reverse coma in acute episodes of metabolic imbalance. Rare patients have a thiamine-responsive form of MSUD, and therefore this vitamin should be tried in all patients. Liver replacement is very efficient in severe forms: Liver transplanted patients no longer require protein restricted diet and the risk of metabolic decompensation during catabolic events is abolished.2
ISOVALERIC ACIDEMIA
Isovaleric acidemia was the first condition recognized as an organic acidemia when the “odor of sweaty feet” in an infant with episodic encephalopathy was shown to be caused by isovaleric acid and by a defect in isovaleryl-CoA dehydrogenase, the mitochondrial enzyme that converts isovaleryl-CoA to 3-methylcrotonyl-CoA.1,2
 CLINICAL FINDINGS
CLINICAL FINDINGS
Isovaleric acidemia may present in the newborn period with encephalopathy, metabolic acidosis, and the characteristic odor of sweaty feet; however, it more often presents in infancy or childhood with episodes of encephalopathy, hepatomegaly, and odor, brought on by infections or protein intake. Severe hyperammonemia is rare. If untreated, the condition is often fatal, and mental retardation is common.
 DIAGNOSIS
DIAGNOSIS
Characteristic metabolites on organic acid analysis include isovalerylglycine and 3-hydroxyisovaleric acid; isovaleric acid itself is not detected by most analytic methods. All accumulated isovaleryl-CoA is normally excreted as nontoxic isovalerylglycine, but during infection or after a protein load, the amounts build up and exceed the liver’s capacity to esterify it, and it appears as isovaleric and 3-hydroxyisovaleric acids. Acylcarnitine analysis will identify accumulation of isovalerylcarnitine. The enzyme defect can be demonstrated in many tissues, including leukocytes and cultured fibroblasts.
 GENETICS
GENETICS
The disorder is inherited as an autosomal recessive trait. The gene encoding isovaleryl-CoA dehydrogenase is on chromosome 15 (15q12-15), and several disease-causing mutations have been identified. Prenatal diagnosis can be based on enzyme assay of cultured amniocytes or chorionic villus cells or, in families in which the mutations are known, molecular analysis. Isovalerylglycine is also increased in the amniotic fluid surrounding an affected fetus but is difficult to detect by standard methods.
 TREATMENT
TREATMENT
Acute episodes of acidosis may require treatment with intravenous glucose and bicarbonate or lactate. Long-term treatment involves restricting leucine intake to amounts necessary for normal growth and development and providing exogenous glycine to increase excretion of isovaleryl-CoA as isovalerylglycine. Using carnitine to increase excretion of the carbon skeleton as isovalerylcarnitine is less well established.
PROPIONIC ACIDEMIA
Propionic acidemia was first described, albeit unknowingly, when ketotic hyperglycinemia was noted as a syndrome of mental retardation and episodic ketoacidosis, neutropenia, thrombocytopenia, osteoporosis, and hyperglycinemia induced by protein intake or infection. The discovery that propionic acidemia (or methylmalonic acidemia or mitochondrial beta-ketothiolase deficiency) could cause the condition was made only when methods to examine and identify organic acids became available.
The disorder is due to a defect in propionyl-CoA carboxylase (PCC), a biotin-containing enzyme that converts propionyl-CoA (an intermediate in the oxidation of isoleucine, threonine, valine, and methionine) to D-methylmalonyl-CoA5,6 (see Fig. 137-2).
 CLINICAL FINDINGS
CLINICAL FINDINGS
Like many other organic acidemias, propionic acidemia can present in the neonate as severe and life-threatening ketoacidosis, hyperammonemic encephalopathy, and bone marrow suppression. An equally common and more chronic course presents later in infancy with episodes of vomiting, ketoacidosis brought on by infections, failure to thrive, and osteoporosis severe enough to cause pathological fractures. Acute striatal damage may occur, usually, but not always, during an episode of ketoacidosis. Developmental delay is occasionally present and is probably caused more by newborn hyperammonemia or recurrent illness in infancy than by the disease itself.
 DIAGNOSIS
DIAGNOSIS
Urine organic acid analysis shows large amounts of 3-hydroxypropionic and methylcitric acids, often with propionylglycine, tiglylglycine, and the abnormal ketone bodies made when propionyl-CoA is incorporated instead of acetyl-CoA. Acylcarnitine analysis shows increased propionylcarnitine, and glycine levels in blood and urine are often elevated. The defect in PCC can be demonstrated in many tissues, including leukocytes and cultured fibroblasts.
 GENETICS
GENETICS
The disorder is inherited as an autosomal recessive trait. PCC is a dodecamer of six α and six β subunits, which are encoded on chromosomes 13 and 3, respectively. Prenatal diagnosis of an affected fetus can be made by demonstrating enzyme deficiency in cultured amniocytes or chorionic villus cells, or by demonstrating elevated methylcitric acid in amniotic fluid.
 TREATMENT
TREATMENT
Dietary restriction of protein or propiogenic amino acids to amounts necessary to support normal growth and development is indicated, usually to protein intake of less than 1 g/kg per day. Treatment with carnitine is useful in secondary carnitine deficiency and may increase excretion of propionyl-CoA as propionylcarnitine. Attacks of ketoacidosis that complicate infections should be treated with fluid and electrolytes. Biotin is usually tried but is rarely effective. Therapy often reduces the frequency and severity of attacks of ketoacidosis, and some patients treated in this manner do well and have normal intelligence; most, however, die in early childhood.
METHYLMALONIC ACIDEMIAS
Accumulation of methylmalonic acid in inherited methylmalonic acidemias is due to deficiency of methylmalonyl-CoA mutase, an adenosyl-B12–requiring enzyme that converts L-methylmalonyl-CoA to succinyl-CoA (see Fig. 137-2). In some patients, mutase deficiency is caused by a defect in the mutase gene, but in others it is caused by a defect in the biosynthesis of adenosyl-B12 from vitamin B12. If this defect also blocks synthesis of methyl-B12, which is required (by N5-methyltetrahydrofolate: homocysteine methyltransferase) for the remethylation of homocysteine to methionine, homocystinuria accompanies accumulation of methylmalonic acid.
Before the enzyme defects were known, patients with defects in B12 biosynthetic were classified by genetic complementation. Table 147-1 shows the enzyme defect responsible for each complementation group. The cblC, cblD, and cblF defects reduce synthesis of adenosyland methyl-B12, and the cblA and cblB defects limit synthesis only of adenosyl-B12.7-10 In addition atypical forms of moderate to mild methylmalonic acid (MMA) excretion have been recently observed in several genetic disorders not directly involved in the main methylmalonate catabolic pathway: Mitochondrial DNA depletion associated with succinyl CoA synthetase complex: SUCLA2 and SUCLG2 (see Chapter 158) in which MMA is associated to succinylcarnitine esters excretion, malonyl-CoA decarboxylase in which mild MMA is associated to huge malonic acid excretion and the still unexplained MAMEL syndrome (Methylmalonic Aciduria Mitochondrial Encephalopathy Leigh like).
 CLINICAL FINDINGS
CLINICAL FINDINGS
As in propionic acidemia, patients with methylmalonic acidemia due to mutase deficiency can present with life-threatening hyperammonemia, ketoacidosis, and thrombocytopenia in the first weeks or months of life; they can also present later, with the more chronic course of ketotic hyperglycinemia, with vomiting, intermittent attacks of life-threatening ketoacidosis, and failure to thrive. As a rule, patients with defects in adenosyl-B12 biosynthesis have somewhat milder disease. Acute striatal damage may occur, often but not always during an episode of ketoacidosis, and late-developing interstitial nephritis is common.
Patients with both homocystinuria and methylmalonic aciduria, however, usually present during the first months of life with failure to thrive; macrocytic anemia; megaloblastosis; nonspecific neurological manifestation such as hypotonia; and, unexpectedly, hemolytic-uremic syndrome. Thromboembolism and episodes of ketoacidosis are rare.
 DIAGNOSIS
DIAGNOSIS
Urine organic acid analysis shows increased methylmalonic acid and, especially in patients with mutase defects, tiglylglycine, 3-hydroxypropionic and methylcitric acids, and the same abnormal ketone bodies found in propionic acidemia. Acylcarnitine analysis shows increased propionylcarnitine, and glycine levels are often elevated in blood and urine. Blood concentrations of vitamin B12 are normal. When homocystinuria is present, amino acid analysis also shows low methionine and high cystathionine; in addition, methylmalonic acid excretion is usually lower than in mutase deficiency. The defect in methylmalonyl-CoA mutase can be demonstrated in many tissues, including leukocytes and cultured fibroblasts, and complementation analysis is available in a few centers.
 GENETICS
GENETICS
All forms of methylmalonic acidemia are inherited as autosomal recessive traits. The gene encoding methylmalonyl-CoA mutase is on chromosome 6 (6p.12-21.1), and several disease-causing mutations have been identified. Prenatal diagnosis of mutase deficiency can be established by performing an enzyme assay on cultured amniocytes or chorionic villus cells; by demonstrating increased methylmalonic and methylcitric acids in amniotic fluid; or, if the mutations in the family are already known, by molecular analysis.
See Chapter 147 for prenatal diagnosis of cbl defects.
 TREATMENT
TREATMENT
Like that of propionic acidemia, treatment of mutase-deficient methylmalonic acidemia relies on restricting dietary protein (or propiogenic amino acids) to amounts necessary to support normal growth and development. Carnitine is useful to treat secondary carnitine deficiency, and attacks of ketoacidosis should be treated with fluid and electrolytes. Some patients treated in this way do well, but most die in early childhood. Liver transplantation may be curative, and successful outcome after combined liver-kidney transplantation in a patient with chronic renal failure has been reported.8 Large doses of vitamin B12 lower methylmalonic acid excretion only in patients with defects of adenosyl-B12 biosynthesis.
Patients with methylmalonic aciduria and homocystinuria are treated with betaine, another methyl donor for the conversion of homocysteine to methionine, and intramuscular vitamin B12. These measures reduce excretion of methylmalonic acid and homocystine, but often without appreciable clinical response. Many patients succumb during childhood because of hemolytic-uremic syndrome or cardiorespiratory arrest, but long-term survival, often with neurological sequelae, can occur.
OTHER RARE ORGANIC ACIDEMIAS
 ISOLATED 3-METHYLCROTONYL-CoA CARBOXYLASE DEFICIENCY
ISOLATED 3-METHYLCROTONYL-CoA CARBOXYLASE DEFICIENCY
The biotin-containing enzyme 3-methylcrotonyl-CoA carboxylase (MCC) converts 3-methylcrotonyl-CoA, an intermediate in leucine oxidation, to 3-methylglutaconyl-CoA. MCC deficiency can exist alone or, in multiple carboxylase deficiency, together with deficiency of carboxylases for propionyl-CoA, pyruvate, and acetyl-CoA.11-13
 CLINICAL FINDINGS
CLINICAL FINDINGS
Many, perhaps most, enzyme-deficient individuals do not develop symptoms.10 Some, however, present between the ages of 1 and 3 years with an infection-induced episode of vomiting, hypoglycemia, hepatomegaly (due to fatty infiltration), hyperammonemic encephalopathy, and hypotonia. Earlier and later presentations have also been described.
 DIAGNOSIS
DIAGNOSIS
Urine organic acid analysis shows increased 3-hydroxyisovaleric acid and 3-methylcrotonylglycine, but the amounts of the latter can be small and hard to detect. Tandem mass spectrometry (MS-MS) shows increased 3-hydroxyisovalerylcarnitine. Serum carnitine levels may be very low, probably as a consequence of 3-hydroxyisovalerylcarnitine excretion. MCC deficiency can be demonstrated in several tissues, including leukocytes and cultured fibroblasts.
 GENETICS
GENETICS
Isolated MCC deficiency is usually inherited as an autosomal recessive trait, but because MCC contains six alpha and six beta subunits, dominant-negative interactions between normal and mutant subunits occasionally cause symptoms, even in heterozygotes.11 The genes encoding the alpha and beta subunits are on chromosomes 3 and 5, respectively, and several pathogenic mutations have been described. Prenatal diagnosis can probably be established by enzyme assay in cultured amniocytes or chorionic villus cells, or by demonstrating increased 3-methylcrotonylglycine in amniotic fluid.
 TREATMENT
TREATMENT
Acute episodes should be treated with fluids, glucose, and electrolytes, and long-term treatment relies on modest protein (or leucine) restriction and oral carnitine to correct carnitine deficiency. Biotin is usually tried but, except in symptomatic heterozygotes, does not reduce the organic acid excretion. Acute episodes may be fatal, but if irreversible brain damage does not occur, the ultimate prognosis is quite good.
3-METHYLGLUTACONIC ACIDURIAS
3-Methylglutaconic aciduria type I (3-MGA type I) is a rare disorder identified in patients who presented a wide spectrum of neurological symptoms ranging from no symptom at 2 years14 to mild developmental delay, severe encephalopathy with basal ganglia involvement, quadriplegia, athetoid movement disorder and leukoencephalopathy in a 61 year old female.
There is a high excretion in urines of 3-methylglutaconic (3-MGG) and 3-methylglutaric which derive from accumulation of 3-MGC CoA proximal to the block. There is also a high excretion of 3-hydroxyisovaleric acid with normal amount of 3-hydroxy-3-methylglutaric acid. Deficient enzyme activity is 3-MGC-CoA hydratase which can be measured in fibroblasts. Several mutations have been identified.
3-MGA type I must be distinguished from many other conditions associated with an abnormal 3-MGG excretion which include Barth Syndrome (3-MGA type II)15 (see Chapter 158), Costeff optic atrophy syndrome (3-MGA type III),16 and disorders of unknown origin summarized as 3-MGA type IV.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


