Eye Disorders
Stacy L. Pineles
Sherwin J. Isenberg
The eye is possibly the fastest developing organ in the body. As soon as 4 to 6 months after birth, some ocular functions are permanently set and if impaired, cannot be fully restored to normalcy. The neonate with a serious ophthalmic disorder may be compared to a time bomb. It is not adequate to simply reverse or cure the immediate problem, since amblyopia may remain. The treatment must be conducted rapidly and effectively, and reconstruction (amblyopia treatment) must be commenced. Thus, the neonatologist has some responsibility to recognize ocular abnormalities and enable early treatment.
▪ GENERAL CONSIDERATIONS
Amblyopia
Amblyopia can be defined as a reduction in vision in the absence of, or beyond that explained by, an apparent organic cause. Amblyopia can be classified as follows: deprivational, strabismic, or refractive. The form of amblyopia that is most feared in infants is deprivation amblyopia. It usually arises before 3 months of age. The cause is blockage of clear images from reaching the retina. It may be unilateral or bilateral and may be caused by anything that obstructs the visual axis, such as a cataract, corneal opacity, or severe eyelid ptosis. There is considerable urgency in reversing this form of amblyopia because good vision can only be attained if the visual axis is cleared within the first 3 to 6 months after birth. This timetable coincides with the “critical period” of ocular development in humans (1). Thus, for example, if a significant unilateral congenital cataract is discovered after 6 months of age, one would not expect excellent visual recovery, even after surgery and optical (usually contact lens or intraocular lens) therapy.
Strabismic amblyopia results from a child preferring one eye when the visual axes are misaligned. Reversal, generally with occlusion of the preferred eye, can only be achieved by age 7 to 9 years; the earlier treatment begins, the better. Refractive amblyopia generally results from significant inequality of the refractive errors in each eye. This form of amblyopia also should be reversed by 7 to 9 years of age, with treatment usually consisting of spectacles (or contact lenses) and, often, occlusion or penalization of the sound eye. Either of these two forms of amblyopia can begin within the first few postnatal months.
Growth and Development of the Eye
At birth, the sagittal (axial) diameter of the eye is 16 mm in full-term neonates (2). It is less in preterm infants at birth. In the first year, this dimension grows 3.8 mm, with half the expected lifetime increase achieved by 12 months of age. With this information, one can appreciate the early anatomic maturity of the eye.
The corneal diameter is often used as an indicator of the size of the entire eye. At term, the corneal diameter averages 10.0 mm. Microcornea (<9 mm) or megalocornea (>11 mm) (Fig. 50.1) suggests a similar abnormality in size of the entire globe, which should lead to an appropriate workup (see following). Ultrasonography can be utilized to determine precisely the size of the entire eye. The tactile corneal reflex is absent in 90% of infants at birth but develops in all by 3 months of age (3).
Three developmental markers exist in the preterm eye that can help the neonatologist define a neonate’s postconceptional age. The tunica vasculosa lentis is a temporary plexus of vessels, which is visible prior to 32 to 34 weeks postconception, crossing the pupil anterior to the lens. The lens surface is covered by these vessels at 27 to 28 weeks postconception, at which time they begin to regress (4). Except for a few vessels at the lens periphery, they should be fully regressed by 34 weeks (Fig. 50.2).
The status of the pupil follows a relatively predictable developmental pattern (Fig. 50.3) (5). At 26 to 31 weeks postconception, the pupillary diameter in relative darkness is quite large (up to 5.0 mm), and the pupil does not respond to light. By 31 weeks, the pupil diameter has decreased to a stable size of 3.5 mm, and the pupil begins to react to light. The light reaction increases in magnitude until reaching stability at term.
The appearance of the macula in the retina is easily appreciated with the ophthalmoscope after the pupil is dilated. The examiner can assess the infant’s postconceptional age by observing the development of three landmarks in the macula: pigmentation, annular reflex, and foveola (Table 50.1) (6). With these three anatomic findings, a neonate’s postconceptional age can be estimated from 27 weeks postconception to term.
▪ EXAMINATION TECHNIQUES
Visual Acuity
In the neonatal period, there is seldom a reason to even attempt to determine the infant’s visual acuity. In the first 2 months after birth, the visual acuity is no better than 20/400 because of immaturity of the retina. The retinal periphery, however, can be stimulated with horizontal optokinetic targets to produce nystagmus. This will prove that the infant is developing some vision. Vertical nystagmus responses develop later. Laboratory techniques can be utilized, if necessary, to more precisely determine the visual acuity. These techniques include preferential forced looking, pattern electroretinograms, and visual-evoked potential. A blink response to light confirms the presence of light perception.
Anterior Segment
The anterior segment can be examined using a strong penlight, with magnification as provided with loupes. Alternatively, a direct ophthalmoscope with a setting of about +5 can be utilized. The examiner should observe the eyelids, conjunctiva, cornea, iris, and lens. The corneal diameter can be measured. As described previously, the pupil diameter initially should be observed with a dim light, followed by evaluation of the reactivity to a bright light.
Posterior Segment
Prior to examining the vitreous and retina, it is usually necessary to dilate the pupil. The choice of dilating agents is important because retinal examinations often are indicated in low-weight preterm infants to rule out retinopathy of prematurity (ROP). Sympathomimetic eye drops can raise a low-weight neonate’s blood pressure (7), whereas anticholinergic eye drops that are thought to be of low concentration can significantly increase gastric acid (8). A safe and effective combination mydriatic eye drop, which is commercially available, is 1.0% phenylephrine/0.2% cyclopentolate. One drop should be applied to both eyes and then repeated 5 to 10 minutes later. A third set occasionally may be required if the iris is darkly pigmented.
Although the eyelids can be held open by an assistant if the examination will be brief, usually an eyelid speculum specifically designed for neonatal use is utilized after application of an anesthetic eye drop. When the examiner is manipulating the eye, infants have been shown to display an oculocardiac reflex, defined as any dysrhythmia or a bradycardia of 10% or more, in as many as 31% of cases (9). Therefore, the assistant should monitor the baby,
as well as the eye, as the retinal examination progresses. During the retinal examination, the cornea tends to become dry and opacify somewhat because of the heat of the light, exposure, evaporation, and reduced tear production especially in preterm infants (10). Thus, while ensuring the stability of the speculum, the assistant also will need to lubricate the cornea.
as well as the eye, as the retinal examination progresses. During the retinal examination, the cornea tends to become dry and opacify somewhat because of the heat of the light, exposure, evaporation, and reduced tear production especially in preterm infants (10). Thus, while ensuring the stability of the speculum, the assistant also will need to lubricate the cornea.
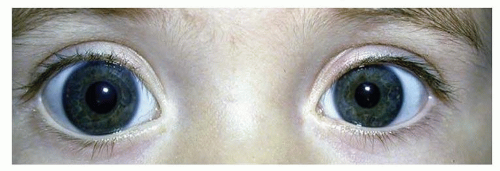 FIGURE 50.1 When the corneal diameter exceeds 11 mm in a neonate, a differential diagnosis must be considered. |
▪ CONGENITAL ANOMALIES
Ocular Size and Shape
Enlarged Eyes
An enlarged eye is suspected when the corneal diameter exceeds 11.0 mm in a term newborn. For confirmation, an A-scan ultrasound can be obtained easily to measure the ocular axial length, which is normally 16 mm at birth (2). If the eye is enlarged, infantile glaucoma caused by an elevated intraocular pressure should be suspected immediately. Infantile glaucoma also often will present with tearing, squinting, photosensitivity, and a cloudy cornea (Fig. 50.4). The cornea often is found to have horizontal lines called Haab striae, which result from a disruption of Descemet membrane. The optic nerve is noted to have an enlarged cup on fundus examination. To differentiate the tearing of glaucoma from that of the much more common nasolacrimal duct obstruction, the examiner should look at, or into, the nostrils. If tears emanate from the nostrils, the nasolacrimal apparatus is patent and glaucoma is possible. If no tears are found in the nostril, a nasolacrimal duct obstruction is more likely.
The treatment of glaucoma is fairly urgent because uncontrolled infantile glaucoma will cause opacification of the cornea, enlargement of the eye, creation of significant myopia, and damage to the optic nerve. If unilateral, the myopia engendered can cause amblyopia, even if the cornea is fairly clear. The corneal opacification can cause deprivation amblyopia.
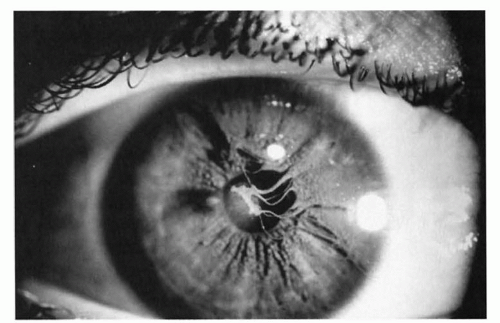 FIGURE 50.2 Persistent pupillary membranes, remaining from tunica vasculosa lentis vessels, are the most common embryonic remnants found in adults. |
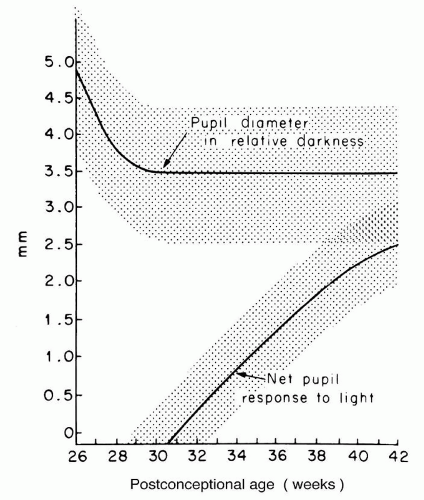 FIGURE 50.3 Diameter of the pupil (mean ± standard deviation) in term and preterm neonates in relative darkness (<10 feet-c) and after light stimulation (600 feet-c). From Isenberg SJ. Examination methods. In: Isenberg SJ, ed. The eye in infancy, 2nd ed. St. Louis, MO: Mosby-Year Book, 1994:47, with permission. |
The infant must be examined while under anesthesia to confirm the diagnosis. After confirmation, the treatment is surgical. The ophthalmologist must open the trabecular meshwork filtration system either internally (goniotomy) or externally (trabeculotomy). If those approaches fail, the ophthalmologist may create an external filtration area (trabeculectomy) or implant an artificial drainage device.
Infantile glaucoma has been associated with other ocular problems, such as aniridia; goniodysgeneses (or mesodermal dysgeneses), which include Axenfeld and Rieger syndromes; and persistent fetal vasculature (see below) and following cataract surgery. It has been associated with a number of systemic disorders and syndromes, including Sturge-Weber, neurofibromatosis, Marfan, Pierre Robin, homocystinuria, Lowe, rubella, Rubinstein-Taybi, and chromosomal abnormalities.
The cornea also may be enlarged on a structural basis without glaucoma. However, in this case, the rest of the eye has a normal shape, as can be demonstrated by ultrasonographic examination and a normal intraocular pressure. Megalocornea is uncommon and usually has an X-linked inheritance pattern.
TABLE 50.1 Development of the Macula | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
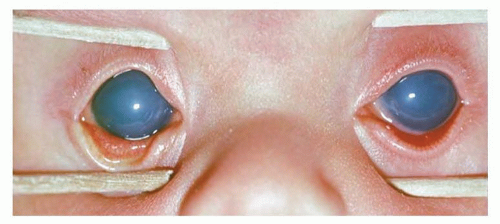 FIGURE 50.4 This cornea is diffusely opacified and enlarged from infantile glaucoma. |
Small Eyes
A small eye will present with a corneal diameter less than 9 mm in a term birth. Confirmation of an axial length less than the normal 16 mm, as shown by ultrasound, is desirable. Microphthalmos can range from an eye that is slightly smaller than normal but otherwise intact to an eye that is so small that it cannot be found on routine examination (anophthalmos). In cases of anophthalmos, a small, often cystic, eye sometimes can be demonstrated by MRI, CT, or ultrasound. It may be associated with Klinefelter syndrome or trisomy 13.
There are two, not infrequent, ophthalmic disorders associated with microphthalmos. A coloboma is a developmental gap generally located inferiorly in the eye. It can be recognized externally as an inferior notch in the pupil caused by missing iris tissue (“keyhole pupil”), which by itself does not affect vision. More ominously, the defect also can include the optic nerve, macula, and other parts of the retina, which can result in legal blindness (Fig. 50.5). Ophthalmologists are now recognizing the frequent combination of systemic findings associated with CHARGE syndrome, consisting of coloboma (C), heart defects (H), atresia choanae (A), retarded growth and development (R), genital hypoplasia (G), and ear anomalies and deafness (E). Facial palsy also is common. At least four of these findings must be present to make the diagnosis.
A second ophthalmic disorder commonly associated with microphthalmos is persistent fetal vasculature, previously known as persistent hyperplastic primary vitreous (11). This disorder often is associated with microphthalmos and hypoplasia of the fovea, as it represents an arrest of ocular development. Many of the sequelae result from abnormal vessels in the vitreous, anterior lens, and equator, causing persistent pupillary membranes, pigmented star-shaped structures on the anterior lens capsule, fibrovascular remnants on the optic nerve (Bergmeister papilla), and nonattachment of the retina. Serious secondary events can ensue, including cataract, glaucoma, lens subluxation, corneal opacities, intraocular hemorrhages, retinal detachments, and chronic inflammation. Because some of these manifestations are treatable, the neonatologist should seek an ophthalmic consultation for any infant with a small eye.
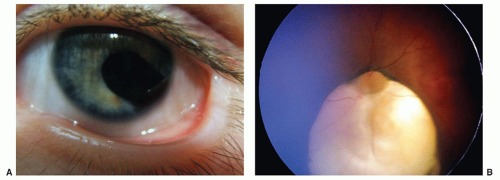 FIGURE 50.5 A: The inferior iris defect, which resembles a keyhole, does not by itself affect vision. B: In this coloboma of the posterior pole of the fundus, the optic nerve is seen at the top. A defect as large as this will compromise vision, especially if fibers to the macula are deficient. |
Eyelid Abnormalities
Congenital eyelid ptosis is readily apparent to the parents and all who observe a baby. An early referral to an ophthalmologist should be made. Appropriate timing of surgical correction is important. If the ptosis threatens the infant’s vision, it should be corrected early—even within the first few postnatal months. The vision can be threatened in two ways. If the ptosis is total or near total, the visual axis will be obstructed and the child may develop deprivation amblyopia. This is unusual because ptosis is seldom total, which allows the child, once head control is established, to elevate the chin to see under the ptotic eyelid. A more likely mechanism of potential visual loss is by an astigmatism induced by the ptotic eyelid applying subtle pressure to the cornea. This unilateral astigmatism can cause refractive amblyopia, even in a young infant. If vision is not threatened, surgery can be deferred until 4 or 5 years of age.
A number of eyelid tumors can present at birth. Most frequent is the capillary hemangioma (Fig. 50.6), which generally continues to grow after birth. The skin overlying the mass can be dimpled and red, resembling a strawberry, or it can assume a diffuse purple color if the lesion is deeper. The lesion does not transilluminate and feels spongy. Left untreated, most of these tumors will spontaneously involute after 1 to 2 years of age. Treatment is indicated if vision is threatened by obstruction of the visual axis or induction of astigmatism, as noted previously. One treatment option is injection of a combination of corticosteroids directly into the tumor or by systemic corticosteroids. The direct route may be safer. Favorable results recently have been reported with oral propranolol (12,13). Dramatic responses have been seen in children even after a few days of oral propranolol treatment. However, the use of propranolol requires monitoring for systemic side effects such as bradycardia, hypoglycemia, and respiratory distress. Therefore, the risks and benefits of all treatment options must be weighed and individualized for each patient. One other treatment option that can be effective in the more superficial tumors is the topical application of timolol drops or gel to the skin overlying the mass. Lymphangiomata and dermoid cysts of the eyelid also can present at birth.
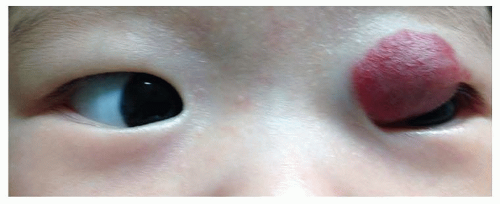 FIGURE 50.6 A hemangioma of the eyelid may not affect vision, even if it is large. |
Corneal Opacities
A number of congenital anomalies can cause corneal opacities at birth. Congenital glaucoma and persistent fetal vasculature have been discussed and must always be ruled out; however, other diagnoses also should be considered.
Birth trauma, usually induced by a forceps placed at or near the eye during delivery, can cause opacities of the cornea. These opacities usually clear within a few days but may leave corneal scars. The corneal damage can leave scarring in the visual axis or induce significant refractive error, which can result in poor vision.
Sclerocornea is a nonprogressive, usually bilateral, anomaly in which the cornea is replaced by opaque sclera-like tissue. Central or total sclerocornea usually is devastating to a child’s vision.
Dermoid tumors of the cornea may affect vision similarly if located centrally. A peripheral corneal tumor can affect vision by inducing refractive error. The tumor may be isolated or part of Goldenhar syndrome.
Peters anomaly, characterized by a central corneal opacity and variable iris-corneal or lens-corneal adhesions, is uncommon but a frequent reason for corneal transplant in infants (Fig. 50.7). The periphery of the cornea usually is normal. It has been associated with fetal alcohol syndrome. Corneal surgery during infancy often is reserved for bilateral cases because the prognosis for good vision following even initially successful corneal transplantation is guarded. In unilateral cases, poor vision is almost inevitable because of amblyopia in the affected eye. The amblyopia may result from a number of causes, including a possible graft rejection, significant refractive errors, recurrent opacities, and secondary glaucoma.
Aniridia
Aniridia, in which much or even the entire iris visible to the examiner is missing, can be compatible with good vision (Fig. 50.8). However, vision can be quite compromised by other ocular associations, including cataracts, peripheral corneal opacities, foveal hypoplasia, glaucoma, and nystagmus. If the vision is quite reduced in infancy, nystagmus usually is found, especially in bilateral cases.
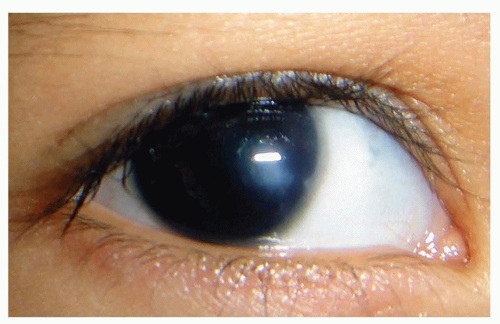 FIGURE 50.7 In this unilateral case of Peters anomaly, the cornea is opaque centrally and clear peripherally. Courtesy of Dr. Federico Velez. |
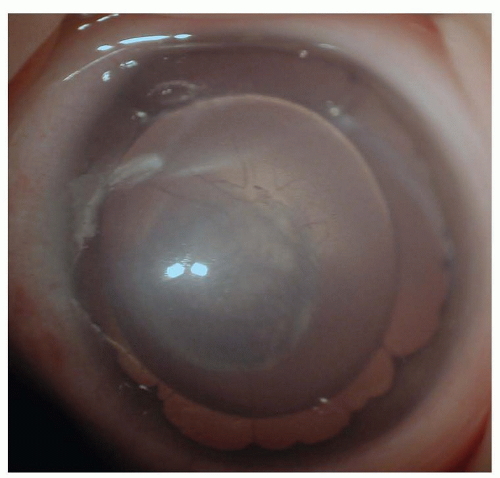 FIGURE 50.8 Aniridia is evident by the largely missing iris. Courtesy of Dr. Federico Velez. |
These children must be followed closely because some of the problems, such as glaucoma and central corneal pannus, may arise later in childhood. The glaucoma is particularly difficult to treat because it often is refractory to medical management. Surgical treatment of the glaucoma can induce a cataract because there may be no iris to protect the lens during and after the operation.
Wilms tumor has been reported to occur in up to one-third of all sporadic aniridia cases. Therefore, periodic abdominal ultrasonography of children with aniridia is justified. An 11p deletion has been associated with the complex of aniridia, ambiguous genitalia, and mental retardation. Wilms tumor also may occur with this deletion.
Cataracts
Lens opacities in infants may be isolated or associated with a systemic condition. The morphology of infantile cataracts often is distinctive, which differentiates the infantile from other forms of cataract (Fig. 50.9). The location of the opacity within the baby’s lens permits a classification of polar, zonular (or lamellar), nuclear, sutural, or total cataract.
About 25% of infantile cataracts are hereditary, especially if bilateral. Therefore, in the workup of infantile cataracts, it is important to examine the parents and siblings. If an asymptomatic cataract that resembles an infantile cataract is found in a family member, the etiology is attributed to heredity, and an extensive workup can be avoided. The most frequent mode of inheritance is autosomal dominant with variable expressivity but almost complete penetrance.
Metabolic problems have been noted to cause cataracts in infants. Among these are hypoglycemia, mannosidosis, hypoparathyroidism, maternal diabetes, and galactosemia. Galactosemia, inherited as an autosomal recessive trait, should be diagnosed on neonatal screening of urinary-reducing substances. But a child with a galactosemia cataract may still present to the pediatrician if he or she was born abroad or has the galactokinase deficiency type, which usually presents after 5 months of age. The cataract resembles a typical oil droplet. Early intervention with a lactose-free diet may reduce the lens level of dulcitol and reverse some or all of the lens opacity. Medications, such as corticosteroids, may cause cataracts but seldom in neonates.
 FIGURE 50.9 A: This zonular (or lamellar) lens opacity occupies a central area of the lens with small satellite opacities. B: The red retinal light reflex is disrupted by a cataract. |
A number of systemic conditions are associated with cataracts. In rubella, cataracts are characteristically total or near-total opacities in a smaller-than-normal lens. In addition, the eye in rubella often is small, has abnormalities of the retinal pigment epithelium (noted as “salt and pepper” changes), and may be glaucomatous. Live rubella virus can survive in the lens for years. Therefore, at surgery, care should be taken with the lens aspirates, especially if personnel in the operating room may be pregnant. Cataracts have been described in other congenital infections, including herpes simplex and varicella.
The presence of a cataract should initiate an appropriate workup with the many causes and associations in mind. The list of other conditions associated with infantile cataract is very long and has been discussed elsewhere (14). To rule out familial cataract, both a family history, including any consanguinity, and an examination of the lenses of the parents and siblings should be undertaken. The history should include questions regarding low-birth weight, ROP, hypoglycemia, serum calcium abnormalities, syndromes, or any systemic disorders. A maternal history of infections while pregnant, diabetes, drug ingestion, and toxin exposure should be sought. Laboratory evaluation should include serum for glucose, blood urea nitrogen, calcium, phosphorus, galactose, and “TORCH” titers (toxoplasmosis, rubella, cytomegalovirus, varicella, and herpes simplex). Lymphocytic choriomeningitis virus may also be responsible for some cataracts in families exposed to rodents (15). Urine should be sent for amino acid levels and hematuria. Other tests should be ordered as indicated by the nonocular findings.
An ophthalmologist should judge if the cataracts are vision threatening. If so and if the child is less than about 4 months old in unilateral cases or 4 to 6 months old in bilateral cases, surgery is urgent to avoid legal blindness from deprivation amblyopia. The surgeon will remove the cataract using an intraocular suction-cutting device (lensectomy) and remove the anterior vitreous (vitrectomy) to avoid the postoperative development of posterior opacified membranes. It should be emphasized that many of the techniques used for cataract surgery in adults are not applicable to children. Compared with adults, a baby’s sclera is more elastic, which can allow an eye to collapse during surgery; the lens itself is softer; the lens capsule is stiffer; and the vitreous is more solid. For these reasons, special training and experience are desirable prior to operating on the cataracts of babies.
The end of the surgery is far from the end of the infant’s visual rehabilitation. An optical device must be used to provide focus after loss of the lens. Spectacles could work, but few infants will keep spectacles in place while in the crib or toddling later. Intraocular lenses, as commonly utilized in adults, are currently under investigation for use in babies. Current intraocular lenses have one fixed power (focal length), which the surgeon must choose at the time of surgery. The refractive power of the baby’s eye will decrease up to 8 diopters by 12 months of age and decrease even more later (16




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
