Esophageal Atresia and Tracheoesophageal Fistula
Spencer W. Beasley
Christchurch Hospital, Christchurch, New Zealand.
Esophageal atresia is a congenital abnormality in which the midportion of the esophagus is absent. Its estimated live birth incidence is between 1 in 3,570 and 1 in 4,500. Most patients have an additional abnormal communication between the trachea and lower esophageal segment called a distal tracheoesophageal fistula. The remainder of the patients have either no fistula (“pure atresia”) or a fistula between the trachea and upper esophageal segment (proximal tracheoesophageal fistula). There is a history of maternal polyhydramnios in about 35% of patients with a distal fistula, and in about 95% of patients with no distal fistula. Prematurity is also common. More than one-half the patients with esophageal atresia have other major congenital anomalies, of which congenital heart disease, urinary tract abnormalities, and gastrointestinal tract abnormalities are the most common. Esophageal atresia and tracheoesophageal fistula are correctable surgically, with generally good results. The diagnosis should be suspected in any newborn infant who appears to have excessive mucus or saliva at birth, with or without respiratory distress.
EMBRYOLOGY
Abnormal Development of the Esophagus
The esophagus develops from the primitive foregut immediately distal to the pharynx. It is likely that the insult that causes esophageal atresia occurs before 32 days’ gestation. The morphologic changes that occur during the embryogenesis of esophageal atresia have been well described, but their interpretation and causes are poorly understood. This has led to a variety of theories being proposed (1), but many of them are now of historical interest only.
Esophageal atresia appears to result from aberrations of differential growth rate, cellular differentiation, and apoptosis. The exact timing and location of the apoptosis in the region of the tracheoesophageal septum seems to be critical for normal separation of the trachea and esophagus (2).
It appears that one or more (as yet unconfirmed) factors alter the rate and timing of cell proliferation and differentiation, and of apoptosis in the region of the esophagus and developing lung bud before 34 days’ gestation. There is some evidence to suggest that the notochord may play a pivotal role (3), possibly through the sonic hedgehog-Gli signaling pathway. The Shh -/- and Gli -/- mutant mice develop esophageal atresia, implying that this signaling pathway is important during foregut development (4).
ANATOMIC AND PHYSIOLOGIC CONSIDERATIONS
Anatomic Variations
Esophageal atresia with distal tracheoesophageal fistula is by far the most common type of abnormality (Fig. 65.1). The length of the upper esophageal segment is variable, but usually reaches within 1 cm of the level of the arch of the azygos vein. The length of the upper esophagus can be estimated preoperatively by the length of tube that can be introduced through the mouth into the esophagus, or by air in the upper esophageal segment seen on plain radiography. At operation, the upper esophagus can be identified when the anesthetist introduces a stiff catheter into the esophagus. The lower esophageal segment commences from the posterior wall of the trachea, usually just proximal to the carina. The level of the tracheoesophageal fistula can be assessed by identifying the carinal air shadow, knowing that the distal esophagus extends at least that far superiorly into the mediastinum. Although the upper esophageal segment is relatively thick walled as a result of hypertrophy from obstruction in utero, the lower segment
has a smaller caliber. At thoracotomy, however, the lower segment may be seen to expand with air during assisted ventilation. Vagal fibers coursing over its surface assist in its identification at operation.
has a smaller caliber. At thoracotomy, however, the lower segment may be seen to expand with air during assisted ventilation. Vagal fibers coursing over its surface assist in its identification at operation.
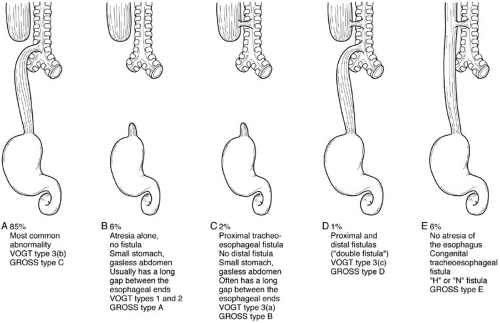 FIGURE 65-1. Types of esophageal atresia. (A) Esophageal atresia and distal tracheoesophageal fistula. (B) Esophageal atresia alone (no fistula). (C) Esophageal atresia with proximal distal fistula. (D) Esophageal atresia with double fistula. (E) “H” fistula—percentage reflects approximate incidence. |
The most important varieties of esophageal atresia and tracheoesophageal fistula are summarized in Fig. 65-1. Various systems of classification have been used, of which the Vogt (1929) and Gross (1953) systems have been the most popular, but do not accommodate all the variations observed (5). Confusion with other classifications and difficulties in their application to unusual variants have gradually led to their abandonment in favor of descriptive terms.
Surgical Approach
The approach employed for any thoracotomy in a neonate is determined by the need for good exposure, as well as the effect the incision will have on subsequent growth and function of the chest wall. An intercostal approach offers little morbidity and satisfactory exposure. Rib resection is no longer employed because of the deformity it may produce, and the posterior fibers of serratus anterior are either retracted anteriorly or divided low at their origin off the chest wall to preserve their innervation.
Many years ago, multiple thoracotomies were performed deliberately as part of staged repairs and in the management of major esophageal complications. It is now evident that multiple thoracotomies increase the likelihood and severity of anterior chest wall deformity, scoliosis, decreased total lung capacity, and decreased vital capacity. In part, this may be a consequence of the reasons for which multiple thoracotomies were performed, such as anastomotic dehiscence or empyema (6). Improvements in neonatal care and surgical technique have meant that staged procedures for repair of esophageal atresia are now performed rarely.
Some surgeons repair esophageal atresia using a thoracoscopic minimally invasive technique (7). To date, thoracoscopy has involved a transpleural approach, and provides a magnified view of the structures in situ. It is not anticipated that it will produce any long-term adverse effects on chest wall development.
Vascular Supply of the Esophagus and Its Influence of Esophageal Mobilization
The cervical portion of the esophagus is supplied by the inferior thyroid artery, which gives off esophageal branches. These branches run vertically downward to the level of the arch of the aorta, where, in the normal esophagus, they anastomose with esophageal branches that come directly from the aorta and bronchial arteries. The remainder of
the thoracic esophagus, particularly that part inferior to the tracheal bifurcation, is supplied by segmental branches from the aorta, but these are of relatively small caliber. They form anastomoses with adjacent vessels, including branches from the intercostal arteries. The distal esophagus is supplied by the ascending branch of the left gastric artery, with some assistance from branches of the inferior phrenic artery. In esophageal atresia, the blood supply to the esophagus is believed to follow the same pattern.
the thoracic esophagus, particularly that part inferior to the tracheal bifurcation, is supplied by segmental branches from the aorta, but these are of relatively small caliber. They form anastomoses with adjacent vessels, including branches from the intercostal arteries. The distal esophagus is supplied by the ascending branch of the left gastric artery, with some assistance from branches of the inferior phrenic artery. In esophageal atresia, the blood supply to the esophagus is believed to follow the same pattern.
The surgical significance of the vascular supply to the esophagus is that the cervical and abdominal portions are supplied by vessels that run along the esophagus, whereas the thoracic portion is supplied segmentally, and thus has the most tenuous connections. There is a risk that excessive mobilization of the thoracic esophagus may render it ischemic. In esophageal atresia with an extensive gap between the two esophageal segments (so-called “long-gap” esophageal atresia), an anastomosis may only be achievable after mobilization of both segments. Knowledge of the vascular anatomy enables the surgeons to be confident of the blood supply of the upper esophageal segment. Even when its mobilization is extensive and continues well up into the neck, there is little risk of it becoming ischemic. However, extensive mobilization of the lower esophageal segment may disrupt its segmental supply and devascularize it. This influences the approach to long-gap esophageal atresia: The lower segment is mobilized only as much as is required to achieve an end-to-end anastomosis without excessive tension.
Circular or spiral esophageal myotomies may further compromise the blood supply of the esophagus, which explains their high complication rate. They may also compromise the innervation of the esophagus, adversely affecting its motility. There are few situations where myotomies are indicated.
Innervation of the Esophagus
The esophagus is supplied by the autonomic nervous system. The sympathetic supply comes from preganglionic neurons in the thoracic and upper lumbar spinal cord. Postganglionic fibers enter the esophagus by way of visceral branches of the sympathetic trunks and greater splanchnic nerves.
Parasympathetic neurons located in the nuclei of the vagus have preganglionic fibers that pass with the vagus nerves. They synapse with short postganglionic neurons situated within the intramural myenteric and submucosal plexuses. These innervate the smooth muscle and secretory cells. An inherent abnormality of the parasympathetic supply in esophageal atresia and the vulnerability of vagal fibers during surgical dissection and mobilization of the esophagus may be responsible for the abnormalities of esophageal function seen in repaired esophageal atresia.
Esophageal Dysmotility
Esophageal motility is abnormal both before and after repair of esophageal atresia. Evidence of inherent congenital motor dysfunction has come from preoperative manometric studies (including studies in patients with H-type tracheoesophageal fistulae) and from observations in rats with Adriamycin-induced esophageal atresia (8). Postoperative manometric and radiologic studies after repair of esophageal atresia have shown that almost the entire length of the esophagus has abnormal motility, regardless of the extent of dissection or tension at the anastomosis.
The surgical procedure may further adversely affect esophageal motility if the fine vagal fibers are injured during mobilization of the esophagus. Esophageal function tends to improve gradually with age, but patients often need to drink with their meals, even into adulthood. Abnormal esophageal motility may contribute to tracheal aspiration. Poor esophageal clearance allows acidic gastric juice to remain in the lower esophagus for a longer period of time than is normal, which may explain the observation that children with esophageal atresia are more likely to suffer complications of gastroesophageal reflux, particularly esophageal stricture. Anastomotic stricture, esophagitis, recurrent tracheoesophageal fistula, and tracheal instability from tracheomalacia all contribute to disordered esophageal motility.
Gastroesophageal Reflux
Gastroesophageal reflux is common in infants with esophageal atresia. Esophageal dysmotility, poor esophageal clearance, and anastomotic narrowing make gastroesophageal reflux more significant and hazardous in infants with esophageal atresia than in normal infants. Esophageal dilatation alone is not effective as definitive treatment for an esophageal stricture secondary to gastroesophageal reflux because ongoing reflux causes the stricture to recur rapidly, often within weeks. Omeprazole may limit stricture formation and reduce esophagitis. It has even been observed to resolve strictures, often without the need for dilatation (9). Where treatment with omeprazole or other proton pump inhibitors fails, or a stricture redevelops despite dilatation, fundoplication (either laparoscopic or open) is required to control the reflux, at which time further esophageal dilatation (radial balloon dilatation under fluoroscopic control) may be performed. Most strictures resolve after the fundoplication, but dysphagia may persist for many months.
Tracheoesophageal Fistula
A distal tracheoesophageal fistula can compromise an infant in two ways. First, escape of air down the tracheoesophageal fistula into the stomach and beyond results in gaseous distension of the abdomen, causing elevation of the diaphragm (so-called “splinting” of the diaphragm) and
restriction of ventilation (Fig. 65-2a). The neonate depends almost entirely on diaphragmatic movement for effective ventilation because the relatively transverse (rather than oblique) configuration of the neonatal ribs means that intrathoracic volume increases little with intercostal contraction. Clinically, the infant is observed to be in respiratory distress, with tachypnea and abdominal distension.
restriction of ventilation (Fig. 65-2a). The neonate depends almost entirely on diaphragmatic movement for effective ventilation because the relatively transverse (rather than oblique) configuration of the neonatal ribs means that intrathoracic volume increases little with intercostal contraction. Clinically, the infant is observed to be in respiratory distress, with tachypnea and abdominal distension.
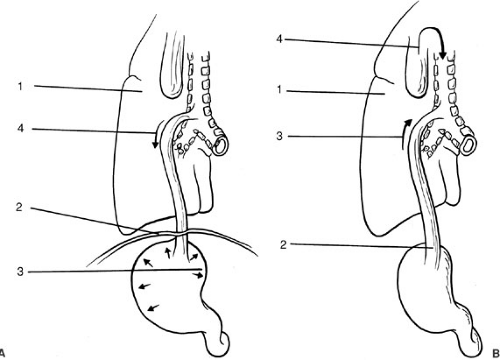 FIGURE 65-2. Physiologic effects of distal tracheoesophageal fistula (A) 1. Hyaline membrane disease may necessitate higher ventilator pressures, which encourage air to pass through the distal fistula. 2. A distended abdomen elevates and “splints” the diaphragm. 3. Gastric distension may result in gastric rupture and pneumoperitoneum. 4. Passage of air through a distal tracheoesophageal fistula diminishes the effective tidal volume. (B) 1. Aspiration of gastric juices leads to soiling of the lungs and pneumonia. 2. Gastroesophageal reflux. 3. Direction of gastric fluid proximally through distal fistula. 4. Overflow of secretions or inadvertent feeding may contribute to aspiration and contamination of the airway. |
Second, in the presence of gastroesophageal reflux (which is common) in esophageal atresia, gastric juice may ascend the esophagus and enter the airway through the fistula (Fig. 65-2b). The frequency with which this mechanism causes contamination of the airway in esophageal atresia is unclear, but in isolated tracheoesophageal fistula (H-fistula), passage of food through the fistula accounts for the recurrent chest infections so often seen. Pneumonia also occurs from overflow of secretions from the blind upper esophageal segment into the airways and by inadvertent feeding before diagnosis. This is the reason infants diagnosed with esophageal atresia must have frequent and regular suctioning of their upper pouch, and must not be fed.
Tracheomalacia
Some degree of structural and functional weakness of the trachea is expected in all infants with esophageal atresia, but in some it may be severe enough to cause respiratory obstruction. There is a deficiency in the tracheal cartilage and an increase in the length of the transverse muscle of the posterior tracheal wall. The cartilage is unable to support the tracheal wall, which has a perimeter greater than normal. The section of the trachea most affected is at the level of the blind-ending proximal esophageal segment. The lower half of the trachea and, occasionally, the whole trachea may be involved. Aberrant vessels (e.g., vascular ring) may increase the severity of tracheomalacia of the adjacent trachea (10). When the abnormality is confined to the intrathoracic trachea, the signs are those of expiratory obstruction. Conditions that increase intrathoracic pressure, such as lower respiratory infection, exacerbate the degree of tracheal collapse. The usual signs of tracheomalacia are those of intermittent expiratory obstruction and normal inspiration. There may be feeding difficulties and vomiting. An esophageal stricture predisposes to inhalation of saliva and food. Distension of the proximal esophagus may compress the trachea and worsen the obstruction of tracheomalacia. In turn, the expiratory obstruction promotes gastroesophageal reflux by increasing intraabdominal pressure. Gastroesophageal reflux, therefore, may be both a cause and result of tracheomalacia.
The natural history of tracheomalacia is one of spontaneous improvement in time. When symptoms are mild, no active intervention is necessary, although the “seal bark” cough may persist into adult life. When tracheomalacia is more severe, careful attention must be paid to feeding. The infant should be offered small amounts of soft foods until late in the first year. Associated gastroesophageal reflux should be managed initially by positioning the head 30 degrees upright and by thickening the feeds. If respiratory symptoms persist, or an esophageal stricture develops, a loose fundoplication should be performed. Aortopexy (tracheopexy) is employed when these measures have failed and the child has recurrent cyanotic episodes due to expiratory obstruction. The rationale for aortopexy relies
on the observation that there are fibrous connections between the posterior surface of the aorta and the anterior wall of the trachea. Drawing the ascending arch of the aorta anteriorly and suturing it to the body of the sternum holds the tracheal lumen open by tightening these fibrous connections.
on the observation that there are fibrous connections between the posterior surface of the aorta and the anterior wall of the trachea. Drawing the ascending arch of the aorta anteriorly and suturing it to the body of the sternum holds the tracheal lumen open by tightening these fibrous connections.
Prematurity
The combination of hyaline membrane disease in premature infants and splinting of the diaphragm from preferential entry of air through the distal tracheoesophageal fistula may produce respiratory embarrassment severe enough to necessitate positive pressure ventilatory support. This may exacerbate the problem of air passing through the fistula, further compromising ventilation. For this reason, management should be directed at maintaining the lowest possible ventilatory pressure to achieve adequate oxygenation, followed by early surgical division of the tracheoesophageal fistula. Because hyaline membrane disease takes 24 to 48 hours to develop, there is time to divide the fistula before the respiratory distress becomes fully established and the complications of inadequate ventilation against an elevated diaphragm or ruptured stomach occur. The earlier practice of performing an emergency gastrostomy alone in infants with major escape of air through a distal tracheoesophageal fistula has been abandoned. This is because the massive air leak often continued after gastrostomy and made ventilation even more ineffective as the air continued to pass preferentially through the fistula.
Closure of the fistula improves the ease of ventilation because
There is no further escape of air down the fistula.
There is no diaphragmatic splinting interfering with ventilation.
Soiling of the lungs with gastric secretions is prevented.
In most infants, esophageal continuity can be achieved at the time of thoracotomy to divide the fistula. If gastric perforation results in a tight pneumoperitoneum, the infant will become extremely difficult to ventilate and will deteriorate rapidly; in this situation, immediate insertion of a wide bore needle into the peritoneal cavity to decompress the abdomen will allow the infant to survive until the fistula is controlled.
DIAGNOSIS OF ESOPHAGEAL ATRESIA
Antenatal Diagnosis
Esophageal atresia may be suspected on antenatal ultrasonography when maternal polyhydramnios, a small stomach, a distended upper esophageal pouch, or abnormal swallowing is observed (11). Diagnostic suspicion is increased when abnormalities known to be associated with esophageal atresia are identified.
Clinical Diagnosis
Any excessively drooling infant should be assumed to have esophageal atresia until proven otherwise. The diagnosis is made when a stiff 10-gauge French catheter introduced through the mouth (Fig. 65-3a) becomes arrested at about 10 cm from the gums. Failure to pass the catheter into the stomach confirms the diagnosis of esophageal atresia. Fluid aspirated up the catheter (saliva) does not usually turn blue litmus pink, as would be expected with gastric aspirate. A tube of smaller caliber may curl up in the proximal pouch and give a misleading impression of esophageal continuity (Fig. 65-3b). A plain radiograph will confirm the tube has not reached the stomach. The tube should not be introduced through the nose because it may injure the nasal passages. Contrast studies, on the rare occasions that they are required, should be performed by an experienced pediatric radiologist, or after transfer to the tertiary institution, and with the use of a small amount (0.5 to 1 mL) of water-soluble contrast. Care must be taken to avoid aspiration.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


