Normal Sexual Differentiation
The normal regulation of sexual differentiation is broadly illustrated in
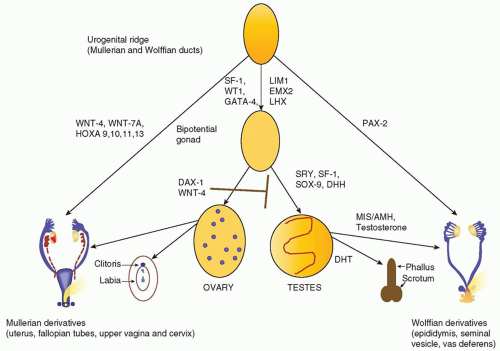
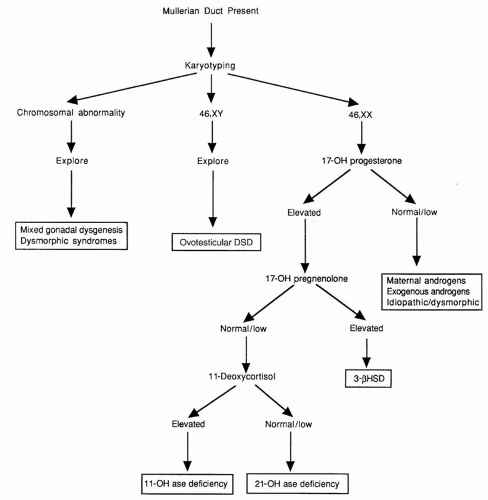
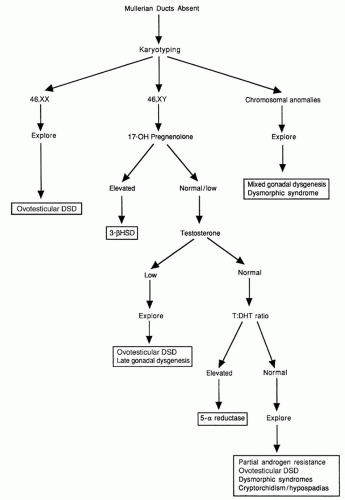
Figure 36.1. All embryos are initially undifferentiated, having a bipotential gonad and the anlagen of both male and female reproductive tracts and genitalia (
1). Differentiation of the gonads as testes or ovaries dictates the subsequent development of the internal and external genitalia. The gonad forms when germ cells migrate from the dorsal endoderm of the yolk sac to populate the genital ridges. At the fifth to sixth week of gestation, these primitive bipotential gonads consist of both cortical (ovarian) and medullary (testicular) components. The genital ridge is composed of three cell types:
Primordial germ cells destined to become prespermatogonia in the male or oogonia in the female
Supporting epithelial somatic cells, destined to become Sertoli cells (male) or granulosa cells (female)
Mesenchymal somatic cells destined to become the steroid-producing Leydig cells (male) or theca cells (female)
Figure 36.1 illustrates the genes involved in the differentiation of the genital ridge into a female or male reproductive tract. WT1 and steroidogenic factor 1 (SF-1) play a role early on in the development of the genital ridge and are critical for gonadal development (
2). Mutations in these two genes are clearly associated with gonadal dysgenesis:
Wilms tumor suppressor gene (WT1) mutations are associated with three related syndromes (the WAGR contiguous gene syndrome, Denys-Drash, and Frasier) that affect renal function and gonadal development (
3).
Mutations in the transcription factor, SF-1, cause agenesis of the adrenal glands and gonads (
4).
Development of the supporting cells as Sertoli or granulosa cells is critical for determining whether the germ cells differentiate as spermatogonia or oogonia. Sexually dimorphic differentiation of the gonads and reproductive system commences when the testis-determining gene is first expressed. In 1959, Ford and colleagues determined that the Y chromosome was necessary for male development (
5); in 1966, the critical region for testis determination was localized to the short arm of the Y chromosome (
6); and in 1990, the primary testis-determining gene was identified at Yp11.3 by positional cloning in patients with 46,XX testicular disorders of sex development (DSD) (
7). This gene, termed SRY (sex-determining region of the Y chromosome), is a member of the SOX family of transcription factors that all contain a high mobility group (HMG) DNA-binding motif (
7). Activation of SRY initiates differentiation of the bipotential gonad as a testis. Loss of function mutations in SRY or a delay in its onset of expression can cause 46,XY complete gonadal dysgenesis, while translocation of SRY to the X chromosome or an autosome causes 46,XX testicular DSD.
SOX9, a presumptive target for SRY, is a related HMG box gene that induces the supporting cells of the gonadal ridge to differentiate as Sertoli cells (
8). SRY and SF-1 work in concert to activate SOX9 gene expression. Inactivating mutations of SOX9 cause campomelic dysplasia, a syndrome of skeletal anomalies and 46,XY gonadal dysgenesis (
9). Mutations affecting genes located downstream of SOX9, SRY, SF-1, and desert hedgehog (Dhh) may also disrupt normal testicular determination (
Fig. 36.1).
Sexually dimorphic differentiation of the wolffian (male anlagen) and mullerian (female anlagen) internal genital tracts depends on the hormonal milieu established by the somatic cells. If SRY is expressed, the primary sex cords develop into testes and the somatic cells differentiate as Sertoli and Leydig cells. SOX9 acts synergistically with WNT1, GATA4, and SF-1 to induce Sertoli cell expression of anti-mullerian hormone (AMH) also known as mullerian inhibiting substance (MIS), a 140-kDa glycoprotein in the TGF-&bgr; family. AMH causes degeneration of the mullerian ducts by inducing apoptotic cell death of the ductal epithelial cells. The Leydig cells secrete testosterone, which stimulates the wolffian ducts to differentiate into the vas deferens, seminal vesicle, and epididymis and virilizes the external genitalia.
Differentiation of the external genitalia requires the activation of testosterone by 5&agr;-reductase-2 to its more active metabolite, dihydrotestosterone (DHT). DHT stimulates fusion of the urethral folds and the labioscrotal swellings to form the corpus spongiosa and scrotum. DHT also stimulates growth of the genital tubercle and prostate. Sexually dimorphic differentiation of the internal ducts and the external genitalia is complete by 12 weeks of gestation. During the latter part of gestation, the testes descend into the scrotum and the phallus enlarges as testosterone production increases under the stimulus of pituitary gonadotropins.
At least two genes, WNT4, a locally secreted signaling glycoprotein, and DAX1, a nuclear hormone receptor in the dosagesensitive sex reversal (DSS) region of the X chromosome, Xp21, are critical for ovarian development. WNT4 prevents Leydig cell differentiation and testosterone production, possibly by suppressing SF-1 activity. DAX1 represses both SF-1 and SOX9 activity and AMH expression (
2). A duplication of either of these genes interferes with normal testicular development to cause a dosagesensitive form of 46,XY gonadal dysgenesis (
4,
10). Furthermore, DAX1 mutations cause adrenal hypoplasia congenita (AHC) and hypogonadotropic hypogonadism, the former is life threatening, and early diagnosis in the first few days of life is critical for prompt institution of treatment with corticosteroid and mineralocorticoid replacement therapy (
11).
In 46,XX embryos, the primary sex cords form follicles by 10 weeks of gestation and the primordial germ cells differentiate as oogonia. Both X chromosomes are needed for oocyte survival. In the absence of one X chromosome, as in Turner syndrome, the ovaries form initially, but degenerate before birth. Fetal ovaries do not secrete AMH; thus, the mullerian ducts differentiate to form the uterus, fallopian tubes, and upper vagina. The absence of testosterone and DHT production by the fetal ovary leads to degeneration of the wolffian ducts and female external genitalia.