Disorders of Glycogen Metabolism
Priya S. Kishnani and Yuan-Tsong Chen
GLYCOGEN STORAGE DISEASE
Glycogen, the storage form of glucose in animal cells, is composed of glucose residues joined in straight chains by α–1,4 linkages and branched at intervals of 4 to 10 residues with α–1,6 linkages. The treelike molecule can have a molecular weight of many millions and may aggregate to form structures recognizable by electron microscopy. 
In skeletal muscle, stored glycogen is a source of fuel that is used for short-term, high-energy consumption during muscle activity; in the brain, the small amount of stored glycogen is used during brief periods of hypoglycemia or hypoxia as an emergency supply of energy. In contrast, the liver takes up glucose from the bloodstream after a meal and stores it as glycogen. When blood glucose levels start to fall, the liver converts glycogen back into glucose and releases it into the blood for use by tissues such as brain and erythrocytes that cannot store significant amounts of glycogen.
Glycogen storage diseases (GSDs) are inherited disorders that affect glycogen metabolism. 
There are more than 12 forms of glycogenoses (see Fig. 154-1). The glycogen found in these disorders is abnormal in quantity or quality, or both. Historically, the glycogen storage diseases were categorized numerically in the order in which the enzymatic defects were identified. This section classifies the diseases by the organs involved and the clinical manifestations.
Liver and muscle have abundant glycogen and are the most commonly and seriously affected tissues. The glycogen storage diseases that principally affect the liver (see Fig. 154-1) typically cause fasting hypoglycemia and hepatomegaly. Some glycogen storage diseases are associated with liver complications like hepatic adenomas with risk for malignant transformation and liver cirrhosis. Other organs besides liver may also be involved; for example, renal dysfunction, cardiac involvement, and myopathy. 
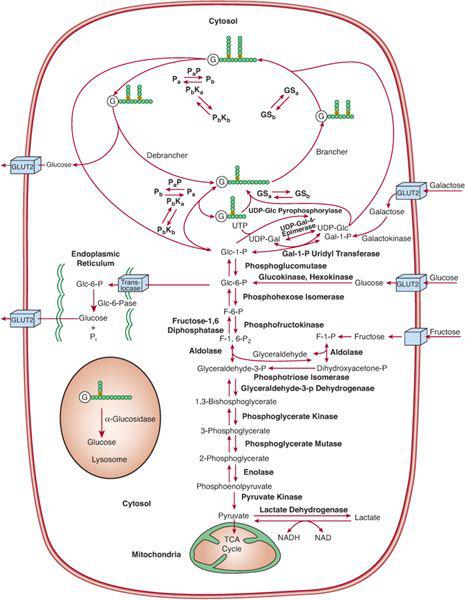
FIGURE 154-1. Metabolic pathways related to glycogen storage diseases and to galactose and fructose disorders. Nonstandard abbreviations are as follows: GSa, active glycogen synthase; GSb, inactive glycogen synthase; Pa, active phosphorylase; Pb, inactive phosphorylase; PaP, phosphorylase a phosphatase; PbKa, active phosphorylase b kinase; PbKb, inactive phosphorylase b kinase; G, glycogenin, the primer protein for glycogen synthesis. (Modified from AR Beaudet. Glycogen storage diseases. In: Isselbacher KJ, et al, eds. Harrison⁽s Principles of Internal Medicine. 13th ed. New York: McGraw-Hill; 1994.)
The role of glycogen in muscle is to provide substrates for the generation of sufficient ATP for muscle contraction. The muscle glycogen storage diseases can be divided into two groups (see Fig. 154-1). The first group is a muscle-energy disorder due to a block in glycolysis and some defects are associated with a compensated hemolysis, suggesting a more generalized defect in glucose metabolism. The second group is characterized by progressive skeletal myopathy and/or cardiomyopathy. 
The overall frequency of all forms of glycogen storage disease is approximately 1 in 10,000 live births. Most of them are inherited as autosomal recessive traits, but phosphoglycerate kinase deficiency and a common form of phosphorylase kinase deficiency are X-linked disorders. The most common childhood disorders are glucose-6-phosphatase deficiency (type I), lysosomal acid α-glucosidase deficiency (type II), debrancher deficiency (type III), and liver phosphorylase kinase deficiency (type IX). The most common adult disorder is myophosphorylase deficiency (type V or McArdle disease) and the late- or adult-onset form of GSD II (Pompe disease). In the past, the prognosis for many glycogen storage diseases was guarded; however, early diagnosis and better management have improved the survival rates, and many affected children are now adults. Consequently, the natural history of these disorders continues to unfold.1
LIVER GLYCOGENOSES
TYPE I GLYCOGEN STORAGE DISEASE (GLUCOSE-6-PHOSPHATASE OR TRANSLOCASE DEFICIENCY, VON GIERKE DISEASE)
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Type I glycogen storage disease is due to a defect in glucose-6-phosphatase in liver, kidney, and intestinal mucosa. It can be divided into two subtypes: type Ia, in which the glucose-6-phosphatase enzyme is defective, and type Ib, which is caused by a defect in the translocase that transports glucose-6-phosphate across the microsomal membrane. Patients with type I disease may develop hypoglycemia and lactic acidosis during the neonatal period but more commonly present at 3 to 4 months of age with hepatomegaly or hypoglycemia. These children often have doll-like faces with fat cheeks, relatively thin extremities, short stature, and a protuberant abdomen that is the result of massive hepatomegaly; the kidneys are enlarged, but the spleen and heart are of normal size.
The biochemical hallmarks of the disease are hypoglycemia, lactic acid acidosis, hyperuricemia, and hyperlipidemia. Hypoglycemia and lactic acidosis can develop after a short fast. Hyperuricemia is present in young children, but gout rarely develops before puberty. Despite hepatomegaly, liver enzymes are usually normal or near normal. Intermittent diarrhea may occur; the mechanism is not clear. In GSD Ib, this could be attributed to inflammation of the bowel. Easy bruising, epistaxis, and menorrhagia are associated with a prolonged bleeding time as a result of impaired platelet aggregation/adhesion.
Hypertriglyceridemia may cause the plasma to appear “milky,” and cholesterol and phospholipid concentrations are also elevated. The lipid abnormality resembles type IV hyperlipidemia.  The hepatocytes are distended by glycogen and fat with large and prominent lipid vacuoles. There is little associated fibrosis.
The hepatocytes are distended by glycogen and fat with large and prominent lipid vacuoles. There is little associated fibrosis.
All these findings apply to both type Ia and Ib glycogen storage diseases, but in type Ib, recurrent bacterial infections due to neutropenia and impaired neutrophil function can occur. Oral and intestinal mucosa ulcerations are common, and inflammatory bowel disease may occur. 
 METABOLIC DERANGEMENT
METABOLIC DERANGEMENT
The block in GSD I is at a critical step, with inability to make glucose via either glycogenolysis or gluconeogenesis. The defects in both type Ia and Ib lead to inadequate conversion of glucose-6-phosphate to glucose in the liver; thus, affected individuals are susceptible to fasting hypoglycemia and a complete dependence on exogenous glucose to maintain normal blood glucose levels.
 GENETICS
GENETICS
Type I glycogen storage disease (OMIM 232200) is an autosomal recessive disorder. The structural gene for glucose-6-phosphatase (type Ia) is located on chromosome 17q21. There are some common mutations repeatedly seen in different ethnic groups, which can be used for genetic testing like R83C (Caucasian 32%, Jewish 93% to 100%); R83H (Chinese 38%); 727 G > T splicing mutation (Japanese 88%, Chinese 36%), 35X, G188R, and Q347X (Caucasian 21%).3
The structural gene for glucose-6-phosphate translocase (type Ib; SLC37A4) is located on chromosome 11q23. A couple common mutations associated with GSD Ib in different ethnic groups are G339C (Caucasian 15%, German 29%) and c.1042delCT (Caucasian 31%, German 32%). 
 DIAGNOSTIC TESTS
DIAGNOSTIC TESTS
Type I glycogen storage disease can be suspected on the basis of clinical presentation and the hallmark laboratory findings of hypoglycemia, lactic acidosis, hyperuricemia, and hyperlipidemia. Neutropenia is noted in GSD Ib patients, typically after the first couple years of life. Administration of glucagon or epinephrine causes little or no rise in blood glucose but increases lactate levels significantly. This test is dangerous during fasting. Gene-based mutation analysis now provides a noninvasive way to diagnosis the disorder for the majority of type Ia and Ib patients—a liver biopsy to demonstrate a deficiency is now rarely required.
 TREATMENT
TREATMENT
Treatment is designed to maintain normal blood glucose levels and is achieved by continuous nasogastric infusion of glucose or oral administration of uncooked cornstarch. Nasogastric drip feeding in early infancy may consist of an elemental enteral formula or may contain only glucose or a glucose polymer to maintain normoglycemia during the night; frequent meals containing a high-carbohydrate content are given during the day.
Uncooked cornstarch acts as a slow-release form of glucose and can be given at a dose of 1.6 g/kg of body weight every 4 hours for children under the age of 2 years. As the child grows older, the cornstarch regimen can be changed to every 6 hours, fructose and galactose intake should be restricted, and dietary supplements of multivitamins, calcium, and vitamin D are required to prevent nutritional deficiencies. Dietary therapy improves glucose control, lactic acidosis, hyperuricemia, hyperlipidemia, and renal function. In some individuals, this therapy fails to completely normalize blood uric acid and lipids levels despite good metabolic control; this is typically noted after puberty. In such situations, hyperuricemia can be controlled with allopurinol. Hyperlipidemia can be managed with lipid-lowering drugs such as HMG-CoA reductase inhibitors and fibrate. Microalbuminuria is an early indicator of renal dysfunction in GSD type I disease and can be treated with low doses of angiotensin-converting enzyme (ACE) inhibitors, such as captopril and lisinopril.  Citrate supplements may be beneficial in preventing or ameliorating nephrocalcinosis and development of urinary calculi.
Citrate supplements may be beneficial in preventing or ameliorating nephrocalcinosis and development of urinary calculi.
In patients with glycogen storage disease type Ib, granulocyte and granulocyte-macrophage colony-stimulating factors (G-CSF) have been used successfully to correct the neutropenia, to decrease the severity of bacterial infection, and to improve the chronic inflammatory bowel disease. Derivatives of 5-amino-salicylic acid (5-ASA) may be a helpful adjunct for treating gastrointestinal complaints. For those patients with GSD-associated inflammatory bowel disease who have not responded to G-CSF and 5-ASA therapy, adalimumab may be an effective therapeutic option.
Some groups advocate orthotopic liver transplantation as a potential cure of type I glycogen storage disease.  Large adenomas (> 2 cm) that increase rapidly in size or number may require partial hepatic resection. Smaller adenomas (< 2 cm) may be treated with percutaneous ethanol injection or transcatheter arterial embolization.4
Large adenomas (> 2 cm) that increase rapidly in size or number may require partial hepatic resection. Smaller adenomas (< 2 cm) may be treated with percutaneous ethanol injection or transcatheter arterial embolization.4
Prior to surgery, the patient’s bleeding status should be evaluated, and good metabolic control should be established. Prolonged bleeding time can be corrected with a constant intravenous glucose infusion for 24 to 48 hours prior to surgery. 
 LONG-TERM COMPLICATIONS
LONG-TERM COMPLICATIONS
Although type I glycogen storage disease mainly affects the liver, multiple organ systems are also involved. Gout usually becomes symptomatic around puberty. Puberty is often delayed; virtually all affected females have ultrasound findings consistent with polycystic ovaries. Despite the ovarian findings, fertility appears to be normal, and there have been several reports of successful pregnancy in this population. Hypertriglyceridemia causes an increased risk of pancreatitis, but premature atherosclerosis has not been documented. 
By the second or third decade, most patients with type I glycogen storage disease develop hepatic adenomas that can hemorrhage and can become malignant. Other complications include iron refractory anemia, pulmonary hypertension, and osteoporosis.
Renal disease is a late complication, and almost all patients above age 20 years have proteinuria. Many affected individuals have hypertension, kidney stones, nephrocalcinosis, and altered creatinine clearance. Glomerular hyperfiltration, increased renal plasma flow, and microalbuminuria can occur before the onset of gross proteinuria. In young patients, hyperfiltration and hyperperfusion may be the only signs of renal abnormalities. With advanced renal disease, focal segmental glomerulosclerosis and interstitial fibrosis are evident on biopsy. 
 PROGNOSIS
PROGNOSIS 
Early diagnosis and effective treatment have improved the outcome; however, renal disease and formation of hepatic adenomas with potential risk for malignant transformation remain as serious complications.
TYPE III GLYCOGEN STORAGE DISEASE (DEBRANCHER DEFICIENCY, LIMIT DEXTRINOSIS)
Type III glycogen storage disease is caused by a deficiency of glycogen debranching enzyme. Debranching and phosphorylase enzyme are responsible for complete degradation of glycogen; when debranching enzyme is defective, glycogen breakdown is incomplete, and an abnormal glycogen that has short outer chains and resembles limit dextrin accumulates.
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Deficiency of glycogen-debranching enzyme causes hepatomegaly, hypoglycemia, short stature, variable skeletal myopathy, and cardiomyopathy. The disorder usually involves both liver and muscle and is termed type IIIa glycogen storage disease. However, in about 15% of patients, the disease appears to involve only the liver and is classified as type IIIb.
During infancy and childhood, the disorder may be at a first glance similar to type I disease, because hepatomegaly, hypoglycemia, hyperlipidemia, and growth retardation are common features of both. However, a more marked elevation of liver enzymes (AST and ALT); an increased creatine kinase in patients with a myopathy; and, especially, an absence of lactic acidosis when fasting are biochemical findings that can separate GSD III from GSD I. Splenomegaly may be present, but kidneys are not enlarged in type III. Remarkably, hepatomegaly and hepatic symptoms in most type III patients improve with age and usually disappear after puberty. However, progressive liver cirrhosis with failure may occur. Hepatocellular carcinoma has also been reported, more typically in patients with progressive liver cirrhosis. The frequency of hepatic adenomas in individuals with GSD III is far less, compared to GSD I.5
In patients with muscle involvement (type IIIa), muscle weakness is usually minimal during childhood but can become severe during the third or fourth decade of life, as evidenced by slowly progressive weakness and muscle wasting in both proximal and distal muscle groups. Electromyographic (EMG) changes are consistent with a widespread myopathy, and nerve conduction may be abnormal. Cardiomyopathy with an asymmetric septal hypertrophy is often noted in individuals with GSD IIIa, but overt cardiac dysfunction is rare. There have been infrequent reports of cardiac conduction abnormalities with resultant death. Myocardial fibrosis has also been reported in a few cases. Hepatic symptoms may be so mild that the diagnosis is not made until adulthood, when neuromuscular disease becomes manifest. Polycystic ovary appears to be a common finding in female patients; fertility, however, does not seem to be affected, and successful pregnancy outcome has been reported in many.
Hypoglycemia, hyperlipidemia, and elevated liver transaminases occur in childhood. In contrast to type I disease, fasting ketosis is prominent, and fasting blood lactate and uric acid concentrations are usually normal. The administration of glucagon 2 hours after a carbohydrate meal causes a normal rise of blood glucose levels, but after an overnight fast, glucagon may provoke no change in blood glucose, the latter timing of administration being potentially dangerous. Serum creatine kinase levels can sometimes be used to identify patients with muscle involvement, but normal levels do not rule out muscle enzyme deficiency.
The histology of the liver is characterized by a universal distention of hepatocytes by glycogen and by the presence of fibrous septa, which is often noted early in the disease. The fibrosis and the paucity of fat distinguish type III from type I GSD. The fibrosis can range from minimal periportal fibrosis to micro-nodular cirrhosis.1
 GENETICS
GENETICS
The type III glycogenoses (OMIM 232400) are inherited as autosomal recessive traits. The disease has been reported in many different ethnic groups, and the frequency is relatively high in non-Ashkenazi Jews of North African descent. The gene for debranching enzyme (AGL) is located on chromosome 1p21. At least 51 different mutations that cause type III disease have been identified in the AGL gene. Two mutations (17delAG and Q6X), both located in exon 3 at amino acid codons 5 and 6, are exclusively found in the subtype GSD IIIb. 
 DIAGNOSTIC TESTS
DIAGNOSTIC TESTS
In glycogen storage disease type IIIa, deficient debranching enzyme activity can be demonstrated in liver, skeletal muscle, and heart. In contrast, type IIIb patients have debranching enzyme deficiency in the liver but not in muscle. In the past, definitive assignment of subtype required enzyme assays in both liver and muscle. DNA-based analyses now provide a noninvasive way of subtyping in the majority of patients.
 TREATMENT
TREATMENT
Dietary management of type III disease is less demanding than in type I. If hypoglycemia is present, frequent high-carbohydrate meals with cornstarch supplements or nocturnal gastric drip feedings are usually effective. A high-protein diet during the daytime plus overnight protein enteral infusion may also be effective in preventing hypoglycemia and, in patients with a myopathy, to prevent endogenous protein breakdown. Exogenous protein can be used as a substrate for gluconeogenesis, a pathway that is intact in type III glycogen storage disease. Aerobic exercise is also very important in individuals with GSD IIIa. Patients do not need to restrict dietary intake of fructose and galactose, as do those with type I disease. Cardiac symptoms are usually minimal and do not require pharmacological intervention; β-adrenergic blockers may be indicated in cases of asymmetric septal hypertrophy and left ventricular outflow tract obstruction. Liver transplantation has been performed in patients with end-stage cirrhosis or hepatic carcinoma.
 PROGNOSIS
PROGNOSIS
Liver symptoms improve with age and usually disappear after puberty. Cirrhosis of the liver may occur later in life, and this needs close monitoring. In type IIIa disease, muscle weakness and atrophy worsen during adulthood.
TYPE IV GLYCOGEN STORAGE DISEASE (BRANCHING ENZYME DEFICIENCY, AMYLOPECTINOSIS, POLYGLUCOSAN DISEASE, OR ANDERSEN DISEASE)
Deficiency of branching enzyme activity results in accumulation of an abnormal glycogen with poor solubility. The disease is referred to as type IV glycogen storage disease, or amylopectinosis, because the abnormal glycogen has fewer branch points, more (1–4) linked glucose units, and longer outer chains, resulting in a structure resembling amylopectin or polyglucosan. 
 CLINICAL PRESENTATION AND LABORATORY FINDINGS
CLINICAL PRESENTATION AND LABORATORY FINDINGS
This disorder is clinically variable. The typical presentation of GSD-IV is characterized by failure to thrive, hepatosplenomegaly, and progressive liver cirrhosis leading to death in early childhood. Patients usually present in the first 18 months of life with hepatosplenomegaly and failure to thrive. The cirrhosis progresses to cause portal hypertension, ascites, esophageal varices, and liver failure that leads to death by age 5 years. A milder, nonprogressive hepatic form is less frequent. 
The neuromuscular presentation of GSD-IV is heterogeneous, and four main variants can be distinguished on the basis of age at onset. The perinatal form presents as fetal akinesia deformation sequence (FADS) and is characterized by multiple congenital contractures (arthrogryposis multiplex congenita), hydrops fetalis, and perinatal death. The congenital form includes hypotonia, muscle wasting, neuronal involvement, inconsistent cardiomyopathy, and death in early infancy. The childhood form is dominated by myopathy or by cardiomyopathy. The adult form can present as an isolated myopathy or as a multisystem disorder with central and peripheral nervous system dysfunction (adult polyglucosan body disease).8,9
Tissue deposition of amylopectin-like materials can be demonstrated in liver, heart, muscle, skin, intestine, brain, spinal cord, and peripheral nerve. The histologic findings in the liver are characterized by both micronodular cirrhosis and faintly stained basophilic inclusions in the hepatocytes.  Definitive diagnosis requires demonstrating that branching enzyme activity is deficient in liver, muscle, cultured skin fibroblasts, or leukocytes, or requires genetic testing of the GBE1 gene.
Definitive diagnosis requires demonstrating that branching enzyme activity is deficient in liver, muscle, cultured skin fibroblasts, or leukocytes, or requires genetic testing of the GBE1 gene.
Definitive diagnosis of the adult disease requires an assay of branching enzyme in leukocytes or nerve biopsy, as the deficiency is limited to those tissues.
 GENETICS
GENETICS
Type IV glycogen storage disease (OMIM 232500) is a rare autosomal recessive disorder. The glycogen-branching enzyme gene is located on chromosome 3p12. All forms of the disease are caused by mutations in the same branching enzyme gene; its characterization in individual patients may be useful in predicting the clinical course.  For the hepatic form, some mutations are associated with a good prognosis and lack of liver disease progression.10 Prenatal diagnosis is available by using cultured amniocytes or chorionic villi to measure the level of enzymatic activity.
For the hepatic form, some mutations are associated with a good prognosis and lack of liver disease progression.10 Prenatal diagnosis is available by using cultured amniocytes or chorionic villi to measure the level of enzymatic activity.
 TREATMENT
TREATMENT
There is no specific treatment for type IV glycogen storage disease. For progressive hepatic failure, liver transplantation has been performed. Caution should be taken in selecting patients for liver transplantation, as a nonprogressive hepatic form of the disease exists. Furthermore, in some patients, extrahepatic manifestations of the disease may occur after transplantation.
TYPE VI GLYCOGEN STORAGE DISEASE (LIVER PHOSPHORYLASE [PGYL] DEFICIENCY, OR HERS DISEASE)
The number of patients with enzymatically documented liver phosphorylase deficiency is small. Regulation of the liver phosphorylase occurs through phosphorylation of the enzyme by phosphorylase kinase, which converts the inactive glycogen phosphorylase b form to the active a form.
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
It appears that patients with liver phosphorylase deficiency typically have a benign course.  These have varied presentations, from hepatomegaly and subclinical hypoglycemia to severe hepatomegaly with recurrent severe hypoglycemia and postprandial lactic acidosis. Patients with the mild phenotype typically present with hepatomegaly and growth retardation early in childhood. Hypoglycemia, hyperlipidemia, and ketosis are usually mild if present. Lactic acid and uric acid concentrations are normal. The heart and skeletal muscles are not involved. The hepatomegaly and growth retardation improve with age and usually disappear around puberty.
These have varied presentations, from hepatomegaly and subclinical hypoglycemia to severe hepatomegaly with recurrent severe hypoglycemia and postprandial lactic acidosis. Patients with the mild phenotype typically present with hepatomegaly and growth retardation early in childhood. Hypoglycemia, hyperlipidemia, and ketosis are usually mild if present. Lactic acid and uric acid concentrations are normal. The heart and skeletal muscles are not involved. The hepatomegaly and growth retardation improve with age and usually disappear around puberty.
 GENETICS
GENETICS
GSD VI (OMIM 232700) is an autosomal recessive disease. Diagnosis rests on enzyme analysis of the liver biopsy or sequencing of the liver phosphorylase gene. 
 TREATMENT AND PROGNOSIS
TREATMENT AND PROGNOSIS
Treatment is symptomatic. A high-carbohydrate diet and frequent meals are effective in preventing hypoglycemia, but most patients require no specific treatment. Overall, long-term prognosis is very good.
TYPE IX GLYCOGEN STORAGE DISEASE (LIVER PHOSPHORYLASE KINASE [PK] DEFICIENCY)
This disorder represents a heterogeneous group of glycogenoses. Phosphorylase, the rate-limiting enzyme of glycogenolysis, is activated by a cascade of enzymatic reactions involving adenylate cyclase, cyclic adenosine monophosphate–dependent protein kinase (protein kinase A), and phosphorylase kinase. Phosphorylase kinase has four subunits (α, β, γ, δ), each encoded by different genes on different chromosomes (X chromosome and autosomes) and differentially expressed in various tissues. The cascade of reactions to activate phosphorylase (from b to a form) is stimulated primarily by glucagon. A glycogenosis could result from an enzyme deficiency along this pathway; the most common is phosphorylase kinase deficiency.
The clinical heterogeneity of type IX GSD results from the complexity of the phosphorylase kinase gene, with its four subunits. Based on the gene/subunit involved, the tissues that are primarily affected, and the mode of inheritance, phosphorylase kinase deficiency can be divided into several subtypes: (1) a benign X-linked recessive liver disease of infancy or childhood, (2) an autosomal recessive liver and muscle disease, (3) a pure myopathy affecting both sexes but predominantly men, (4) an autosomal recessive severe liver disease associated with cirrhosis, and (5) a fetal infantile cardiopathy.
X-LINKED LIVER PHOSPHORYLASE KINASE DEFICIENCY
X-linked liver phosphorylase kinase deficiency (OMIM 306000) is one of the most common liver glycogenoses. The X-linked gene (PHKA2) is located on Xp22.2-p22.1 and encodes the liver isoforms of the α subunit. Defects in PHKA2 give rise to X-linked liver PK deficiency, characterized by hypoglycemia, hepatomegaly, chronic liver disease, growth retardation and delayed motor development, hypercholesterolemia, hypertriglyceridemia, elevated liver enzymes, and hyperketosis after fasting. Lactate and uric acid levels are normal. The rise in blood glucose concentration following the administration of glucagon is normal. Typically, children present between the ages of 1 and 5 years with the above symptoms, which typically ameliorate during puberty. Most adults achieve a normal final height and are practically asymptomatic. 
Liver histology shows glycogen-distended hepatocytes. 
AUTOSOMAL LIVER AND MUSCLE PHOSPHORYLASE KINASE DEFICIENCY
Autosomal liver and muscle phosphorylase kinase deficiency (OMIM 261750) is inherited in an autosomal recessive manner. As in the X-linked form of the disorder, hepatomegaly and growth retardation are the predominant symptoms in early childhood. Some patients also exhibit muscle hypotonia and have reduced activity of phosphorylase kinase in muscle. This form of the phosphorylase kinase deficiency is caused by mutations in the β subunit of the gene PHKB, located on chromosome 16q12-13.
AUTOSOMAL LIVER PHOSPHORYLASE KINASE DEFICIENCY
In contrast to the benign course of X-linked phosphorylase kinase deficiency, patients with this autosomal recessive form of liver phosphorylase kinase deficiency (OMIM 172471) have a more severe clinical course characterized by marked hepatomegaly, recurrent fasting hypoglycemia, impaired glucagon response, and fibrosis that in the majority of reported cases progressed to frank liver cirrhosis and progressive hepatic failure. Hepatic adenomas have been seen in some individuals. This form of the phosphorylase kinase deficiency is due to mutations in the testis/liver isoform of the γ subunit of the gene (γTL, PHKG2) located on chromosome 16p12.1.12
MUSCLE-SPECIFIC PHOSPHORYLASE KINASE DEFICIENCY
Muscle-specific phosphorylase kinase deficiency (OMIM 172470) could be inherited in an X-linked or autosomal recessive manner. The structural gene for the muscle-specific (PHKA1) α subunit is located at Xq12. Defects in PHKA1 result in X-linked deficiency of phosphorylase kinase in muscle. Symptoms include exercise intolerance, cramps, myalgia, weakness, atrophy, and myoglobulinuria. The activity of the enzyme is decreased in muscle but when tested has been normal in liver and blood cells. There is no hepatomegaly or cardiomegaly.13
The gene for the muscle γ subunit (γM, PHKG1) has been localized to chromosome 7p12. No mutations in this gene have been reported so far.
CARDIAC-SPECIFIC PHOSPHORYLASE KINASE DEFICIENCY
Several sporadic cases of cardiac-specific phosphorylase kinase deficiency (OMIM 261740) have been reported. All affected individuals died during infancy from cardiac failure due to massive glycogen deposition in the myocardium. However, recent study showed that the existence of cardiac-specific primary phosphorylase kinase deficiency is questionable, because no mutations in the eight genes encoding the phosphorylase kinase subunits were found.14 Instead, a recurrent activating R531Q mutation in the γ-2 subunit of AMP activated protein kinase (PRKAG2) gene was the cause in most patients. 
 DIAGNOSIS
DIAGNOSIS
The diagnosis of glycogen storage disease type IX is complicated by the highly complex nature of phosphorylase kinase. Diagnosis of phosphorylase kinase deficiency requires the enzymatic defect be present in affected tissues; the diagnosis can be missed without studies of the liver, muscle, or heart in certain instances. Thus, in many instances, mutation analysis is needed. 
 TREATMENT
TREATMENT
The treatment for liver phosphorylase kinase deficiency is based on symptoms. A high-carbohydrate diet and frequent meals are effective in preventing hypoglycemia, but most patients require no specific treatment. Prognosis for the X-linked and certain autosomal forms is good; adult patients have normal stature and minimal hepatomegaly. Individuals with mutations in the γ subunit typically have a more severe clinical course with progressive liver disease. There is no treatment for the fatal form of isolated cardiac phosphorylase kinase deficiency other than heart transplantation.
TYPE 0 GLYCOGEN STORAGE DISEASE (LIVER GLYCOGEN SYNTHASE DEFICIENCY)
Glycogen synthase normally catalyzes the formation of α-1,4-linkages that elongate chains of glucose molecules to form glycogen. In GSD 0, only glycogen synthesis in the liver is impaired.
Strictly speaking, this is not a type of glycogen storage disease, as the deficiency of the enzyme leads to decreased glycogen stores.
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Patients present in early infancy with early morning drowsiness and fatigue, and sometimes convulsions associated with hypoglycemia and hyperketonemia. There is no hepatomegaly, prominent muscle symptoms, or hyperlipidemia. Most children are cognitively and developmentally normal. Short stature and osteopenia are common features, but other long-term complications, common in other types of glycogen storage disease (GSD), have thus far not been reported in GSD 0.
Because a substantial fraction of dietary carbohydrate is normally stored in the liver as glycogen, inability to synthesize hepatic glycogen causes postprandial hyperglycemia after ingesting a carbohydrate-containing meal. Prolonged hyperglycemia and elevation of lactate with normal insulin levels after glucose administration suggest a possible glycogen synthase deficiency. Definitive diagnosis requires a liver biopsy to measure the enzyme activity or to identify mutations in the liver glycogen synthetase gene.
 GENETICS
GENETICS
GSD 0 (OMIM 240600) is caused by mutations in the GYS2 gene located on chromosome 12p12.2 and is inherited in an autosomal recessive manner. 
 TREATMENT
TREATMENT
Treatment is symptomatic and involves frequent meals rich in protein and a nighttime supplement of uncooked cornstarch to alleviate hypoglycemia. Prognosis seems good, as patients survive to adulthood with resolution of hypoglycemia except during pregnancy.17
TYPE XI GLYCOGEN STORAGE DISEASE (HEPATIC GLYCOGENOSIS WITH RENAL FANCONI SYNDROME, FANCONI-BICKEL SYNDROME)
Type XI glycogen storage disease (OMIM 227810) is a rare autosomal recessive disorder caused by defects in the facilitative glucose transporter 2 (GLUT2), which transports glucose in and out of hepatocytes, pancreatic beta cells, and the basolateral membranes of intestinal and renal epithelial cells.18 (GLUT2 is described in Chapter 157.)
MUSCLE GLYCOGENOSES
GLYCOGENOSIS WITH MUSCLE ENERGY IMPAIRMENT
TYPE V GLYCOGEN STORAGE DISEASE (MUSCLE PHOSPHORYLASE DEFICIENCY, OR MCARDLE DISEASE)
Deficiency of muscle phosphorylase is the prototype muscle-energy disorder. Deficiency of this enzyme in muscle limits ATP generation by glycogenolysis and results in glycogen accumulation.
 CLINICAL PRESENTATION AND LABORATORY FINDINGS
CLINICAL PRESENTATION AND LABORATORY FINDINGS
Symptoms usually develop first in adulthood and are characterized by exercise intolerance with muscle cramps. Two types of activity tend to cause symptoms: (1) brief exercise of great intensity, such as sprinting or carrying heavy loads, and (2) less intense but sustained activity, such as climbing stairs or walking uphill. Moderate exercise, such as walking on level ground, can be performed by most patients for long periods. Many patients experience a characteristic “second wind” phenomenon; if they rest briefly at the first sign of muscle pain, they can resume exercise with more ease. Typically, patients experience episodic muscle cramping and pain upon strenuous exercise; about 35% report permanent pain that impacts general activities and sleep. About half report burgundy-colored urine after exercise, the consequence of myoglobinuria secondary to the rhabdomyolysis. Intense myoglobinuria after vigorous exercise may cause renal failure. Although most patients are diagnosed in the second or third decade, many report weakness and lack of endurance since childhood. Later in adult life, persistent weakness and muscle wasting may develop with fatty replacement. In rare cases, EMG findings may suggest an inflammatory myopathy, and the diagnosis can be confused with polymyositis. Rarely, patients present in infancy with progressive weakness, hypotonia, respiratory distress, and early death. This has to be considered in the differential diagnosis of the floppy baby syndrome, but this timing is very rare. Thus, there can be a broad, heterogeneous spectrum of clinical presentation with the neonatal form, which is rapidly fatal at one extreme, with the benign classical form with myalgia, cramps, and dark-colored urine at the other. Some cases have presented as late as the eighth decade of life. Despite disability, longevity does not appear to be affected, except in the neonatal form.
The level of serum creatine kinase is usually elevated at rest and increases more after exercise. Exercise also increases the levels of blood ammonia, inosine, hypoxanthine, and uric acid. The latter abnormalities are attributed to accelerated recycling of muscle purine nucleotides in the face of insufficient ATP production.19
 GENETICS
GENETICS
Type V glycogen storage disease (OMIM 232600) is an autosomal recessive disorder that does not appear to have ethnic predilection. In most cases, heterozygous carriers are clinically unaffected; however, there are reports of heterozygotes manifesting with this disease. The gene for muscle phosphorylase (PYGM) is located on chromosome 11q13. The most common mutation noted in 90% of North American patients is a nonsense mutation that changes an arginine to a stop at codon 49 (R49X), and the most common mutation found in about 60% of Japanese patients is deletion of a single codon (F708). Other common Caucasian mutations (G204S in exon 5 and K542T in exon 14) make DNA-based diagnosis and carrier testing for McArdle disease possible for the two populations. 
 DIAGNOSIS
DIAGNOSIS
Lack of an increase in blood lactate levels and exaggerated blood ammonia elevations after an ischemic exercise test are indicative of muscle glycogenosis and suggest a defect in the conversion of glycogen or glucose to lactate. The abnormal exercise response, however, is not limited to type V disease and can occur with other defects in glycogenolysis or glycolysis, such as deficiencies of muscle phosphofructokinase or debranching enzyme (noted when the test is done after fasting). Definitive diagnosis is made by enzymatic assay in muscle tissue or by mutation analysis of the myophosphorylase gene.21
 TREATMENT
TREATMENT
In general, avoidance of strenuous exercise can prevent the major attack of the rhabdomyolysis. Aerobic training, oral administration of sucrose, or injection of glucagon can augment exercise tolerance. A high-protein diet may increase exercise endurance in some patients. Vitamin B6 supplementation can reduce exercise intolerance and muscle cramps and increase general well-being. Creatine supplementation at a dose of 10 to 20 g daily is thought to improve muscle function  excitability that induces fatigue in both normal individuals and those with McArdle disease.
excitability that induces fatigue in both normal individuals and those with McArdle disease.
In general, avoiding strenuous exercise prevents the symptoms. 
TYPE VII GLYCOGEN STORAGE DISEASE (MUSCLE PHOSPHOFRUCTOKINASE DEFICIENCY, OR TARUI DISEASE)
Type VII disease is caused by a deficiency of muscle phosphofructokinase, which catalyzes the conversion of fructose-6-phosphate to fructose-1,6-diphosphate and is a key regulatory enzyme of glycolysis.
Phosphofructokinase is composed of three isozyme subunits (M, muscle; L, liver; and P, platelet), which are encoded by different genes and are differentially expressed in tissues. Skeletal muscle contains only M-subunit isozymes, and red blood cells contain a hybrid of L and M forms. Type VII disease is due to defective M isoenzyme, which causes complete enzyme deficiency in muscle and partial deficiency in red blood cells.
 CLINICAL PRESENTATION AND LABORATORY FINDINGS
CLINICAL PRESENTATION AND LABORATORY FINDINGS
The features are similar to those in type V disease. However, several features of type VII disease are distinctive: (1) Exercise intolerance is usually evident in childhood, is more severe than in type V disease, and may be associated with nausea and vomiting; (2) a compensated hemolysis occurs as evidenced by an increased level of serum bilirubin and reticulocyte count; (3) hyperuricemia is common and becomes more marked after exercise; (4) an abnormal glycogen-resembling amylopectin is present in muscle fibers; (5) there is no spontaneous second-wind phenomenon; and (6) exercise intolerance is particularly acute following meals rich in carbohydrate. In contrast, patients with type V disease can metabolize glucose derived from either liver glycogenolysis or exogenous glucose. Indeed, glucose infusion improves exercise tolerance in type V patients. 
Rare type VII variants have been reported (see online version). 
 GENETICS
GENETICS
Type VII glycogen storage disease (OMIM 232800) is inherited in an autosomal recessive manner but exhibits a strong male predominance. The disease appears to be rare, and most reported patients are either Japanese or Ashkenazi Jews. The gene for the M isoenzyme is located on chromosome 12q13.3. 
 DIAGNOSIS
DIAGNOSIS
The molecular diagnosis is practical only in limited patient populations or in families with known mutations. For most patients, diagnosis requires biochemical or histochemical demonstration of the enzymatic defect in the muscle. The M-isoenzyme defect must be demonstrated in muscle, red blood cells, or cultured skin fibroblasts.
 TREATMENT
TREATMENT
There is no specific treatment for this condition. 
OTHER MUSCLE GLYCOGENOSES WITH MUSCLE-ENERGY IMPAIRMENT
Five additional enzyme defects produce muscle glycogenoses, namely deficiencies in phosphoglycerate kinase, phosphoglycerate mutase, lactate dehydrogenase, fructose 1,6-bisphosphate aldolase A, and pyruvate kinase. All five enzymes affect terminal glycolysis, and deficiency causes muscle-energy impairment similar to that in type V and VII disease. The failure of blood lactate to increase in response to exercise can be used to separate muscle glycogenoses from disorders of lipid metabolism, such as carnitine palmitoyl transferase II deficiency and very-long-chain acyl-coenzyme-A dehydrogenase deficiency, which also cause muscle cramps and myoglobinuria. Muscle glycogen levels may be normal in the disorders affecting terminal glycolysis, and definitive diagnosis is made by assaying the enzymatic activity in muscle.
GLYCOGENOSES WITH PROGRESSIVE SKELETAL OR CARDIAC MYOPATHY
GLYCOGEN STORAGE DISEASE TYPE II (ACID α-1,4-GLUCOSIDASE DEFICIENCY, ACID MALTASE DEFICIENCY, OR POMPE DISEASE)
Glycogen storage disease type II, or Pompe disease, is caused by a deficiency of lysosomal acid α-1,4-glucosidase (GAA), an enzyme responsible for degrading glycogen in lysosomal vacuoles. Deficiency of GAA results in lysosomal glycogen accumulation in multiple tissues and cell types, with cardiac, skeletal, and smooth muscle cells being the most seriously affected. This disease is thus characterized by accumulation of glycogen in lysosomes as opposed to its accumulation in cytoplasm in the other glycogenoses.
 CLINICAL PRESENTATION AND LABORATORY FINDINGS
CLINICAL PRESENTATION AND LABORATORY FINDINGS
The disorder encompasses a range of pheno-types, each including myopathy but differing in age of onset, organ involvement, and clinical severity. It is broadly categorized into infantile and late-onset forms. The latter encompasses all forms of the disease (juvenile or late-childhood form and adult form). At the most severe end of the disease spectrum is the infantile form with massive cardiomegaly, hypotonia, and death prior to 1 year of age. Infants may appear normal at birth but soon develop generalized muscle weakness with feeding difficulties, macroglossia, hepatomegaly, and congestive heart failure due to a rapidly progressive hypertrophic cardiomyopathy. Electrocardiographic findings include a very suggestive pattern with high-voltage QRS complexes and a shortened PR interval. Death usually occurs from cardio-respiratory failure.
Late-onset Pompe disease is characterized by skeletal muscle manifestations, usually without cardiac involvement, and a more slowly progressive course but with significant morbidity. In the juvenile form, some patients present with delayed motor milestones (if age of onset is early enough) and difficulty in walking, which is followed by swallowing difficulties, proximal muscle weakness, and respiratory muscle involvement; it can cause death before the end of the second decade. These children may have cardiac involvement.
Adults present with a slowly progressive myopathy, typically without cardiac involvement and with onset between the second and seventh decades. The clinical picture is dominated by slowly progressive proximal muscle weakness with truncal involvement and greater involvement of the lower than the upper limbs. Pelvic girdle, paraspinal muscle, and diaphragm are the most seriously affected. With disease progression, patients become confined to wheelchairs and require artificial ventilation. Respiratory symptoms are manifested by somnolence, morning headache, orthopnea, and exertional dyspnea, which eventually lead to sleep-disordered breathing and respiratory failure. Respiratory failure is the cause of significant morbidity and mortality in this form of the disease. In rare instances, respiratory insufficiency with minimal to no muscle weakness are the presenting symptoms. The age of death in late-onset Pompe disease varies from early childhood to late adulthood.
Laboratory findings include elevated levels of serum creatine kinase, aspartate transaminase, and lactate dehydrogenase, particularly in infants. Vacuolated lymphocytes may be observed on a blood smear. Echocardiography may reveal thickening of both ventricles or of the intraventricular septum or may reveal left ventricular outflow tract obstruction. Muscle biopsy shows the presence of vacuoles that stain positively for glycogen, and muscle acid phosphatase is increased. Electron microscopy reveals the glycogen accumulation. EMG reveals myopathic features with irritability of muscle fibers and pseudomyotonic discharges. Serum creatine kinase concentration is not always elevated in adults, and, depending on the muscle biopsied or tested, muscle histology or EMG may not be abnormal. It is prudent to examine affected muscle for enzymology if the clinical diagnosis is suspected.23,24
 GENETICS
GENETICS
Pompe disease (OMIM 232400) is an autosomal recessive pan-ethnic disorder. The infantile-onset form has an apparent higher incidence among African Americans and Chinese, whereas the late-onset adult form has a higher incidence in the Netherlands. The gene for acid-α glucosidase is on chromosome 17q25. More than 100 mutations and numerous variants in the GAA gene have been identified. The mutations are spread across the gene, though missense mutations in exon 14 may be overrepresented. Among the recurrent mutations in the infantile-onset cases is a single base pair deletion, Δ525T, that is seen in 9% of US cases. This same mutation accounts for 34% of Dutch cases. The exon 18 deletion mutation is seen in infantile-onset cases and accounts for about 25% of Dutch and Canadian cases but only about 5% of US cases. The leaky IVS1(-13T->G) splice-site mutation accounts for about 50% of late-onset cases. 
 DIAGNOSIS
DIAGNOSIS
Diagnosis can be established by demonstrating absence or reduced levels of acid-α-glucosidase activity in muscle, cultured skin fibroblasts, and blood cell–based assays such as leukocytes, mononuclear cells, or dried blood spot as used in newborn screening. Deficiency is usually more severe in the infantile form than in late-onset Pompe disease. Prenatal diagnosis using amniocytes or chorionic villi is available. 
 TREATMENT
TREATMENT
Until recently, there was no effective treatment for Pompe disease, and treatment options were limited to supportive or palliative care. Enzyme replacement therapy (ERT) with alglucosidase alfa (Myozyme), which provides recombinant human acid α-glucosidase, is currently available as the first effective treatment for this once-devastating and lethal disease. Data from various clinical trials have shown that recombinant acid α-glucosidase improves cardiac function and skeletal muscle function in individuals with Pompe disease across the disease spectrum. Response of skeletal muscle function has, however been variable. Patients who have a negative response to cross-reacting immunologic material and develop a high titer antibody against the infused enzyme seem to respond to the ERT less favorably. For patients with the late-onset form of the disease, a high-protein diet may be beneficial. Ventilatory support, when indicated, should be used. 
MUSCLE GLYCOGEN SYNTHASE DEFICIENCY
Glycogen storage disease due to muscle glycogen synthase (glycogen synthase I, GYS1) deficiency (OMIM 138570) was recently reported in three children of consanguineous parents of Syrian origin. The oldest brother died from sudden cardiac arrest at age 10.5 years; the younger brother at age 11 showed muscle fatigability, hypertrophic cardiomyopathy, an abnormal heart rate, and hypotension while exercising; and a 2-year-old sister had mildly impaired cardiac function at rest. Muscle biopsies showed lack of glycogen, predominantly oxidative fibers and mitochondrial proliferation. Glucose tolerance was normal. Molecular study revealed a homozygous stop mutation (R462>ter) in the muscle glycogen synthase gene.27
DANON DISEASE AND AMP-ACTIVATED PROTEIN KINASE GAMMA 2 DEFICIENCY
Deficiencies of lysosomal-associated membrane protein 2 (LAMP2, also called Danon disease; OMIM 300257) and AMP-activated protein kinase gamma 2 (PRKAG2) deficiency (OMIM 602743) result in accumulation of glycogen in the heart and skeletal muscle. Clinically, these patients present primarily with hypertrophic cardiomyopathy but can be distinguished from the usual causes of hypertrophic cardiomyopathy due to defects in sarcomere-protein genes by their electrophysiological abnormalities, particularly ventricular pre-excitation and conduction defects.28 The onset of cardiac symptoms, including chest pain, palpitations, syncope, and cardiac arrest, can occur between the ages of 8 and 15 years for LAMP2 deficiency; this is younger than the average age for patients with PRKAG2 deficiency, which is 33 years. The prognosis for LAMP2 deficiency is poor, with progressive end-stage heart failure early in adulthood. By contrast, cardiomyopathy due to PRKAG2 mutations is compatible with long-term survival except for a rare congenital form that presents in early infancy with a rapid fatal course.14
REFERENCES
See references on DVD.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


