Craniofacial Anomalies
Jeffrey L. Marsh
Pediatric Plastic Surgery, Cleft Lip/Palate and Craniofacial Deformities Center, St. John’s Mercy Medical Center, St. Louis, Missouri 63141.
Congenital deformities of the skull and face, or craniofacial anomalies (1), differ from anomalies of other regions of the body in several important ways:
Craniofacial anomalies are usually overt at birth.
Parents and health care professionals alike can recognize an error of in utero craniofacial development without the aid of specialized physical examination or diagnostic medical imaging.
Craniofacial dysmorphology has major psychosocial consequences in most cultures.
In contrast, cavitary anomalies (e.g., intracranial, intrathoracic, or intraabdominal) usually do not present perinatally, generally require ancillary diagnostic maneuvers for definition, and are rarely recognized by the child’s parents or society at large.
The delivery room personnel are often the first to share the shock of a newborn’s craniofacial anomaly with the parents. Although delivery room personnel may take a positive approach in informing the parents, misinformation, fear, and confusion are frequently conveyed. Furthermore, there is a pervasive prejudice within our society that facial deformity implies impaired cognitive function; this assumption is false in most cases. Whereas impaired social integration may not be a paramount concern for parents of a child with congenital defects that are not visible, for parents of a craniofacially deformed infant, how the child will function with peers, get a job, and find a mate are paramount questions that arise shortly after birth.
This chapter intends to be a guide for health care professionals who may be confronted with a child with a craniofacial anomaly and who want to better understand the nature of the anomaly, its physical and psychological consequences, and the ability of contemporary interdisciplinary team care to habilitate affected individuals.
CRANIOFACIAL DEVELOPMENT
Normal Development
The development of the head is a complex process that has been studied in detail over the past century with increased comprehension thanks to technological advances in investigative tools. Excellent reviews of current knowledge and hypotheses have been published (2,3,4). This brief section focuses on the key features of normal development that are believed to be implicated in the expression of craniofacial anomalies.
Fetal and postnatal craniofacial growth and development are well characterized. The cranium and upper face reflect the early maturation of the central nervous system, whereas the middle and lower face require dental eruption and puberty to reach definitive form and size. The term infant is born with one-fourth of the adult brain volume. By age 2 years, three-fourths of the adult brain volume has developed. The eyes follow the brain’s growth pattern and reach almost adult size by age 4 years. This rapid growth is responsible for the dominance of the cranium and prominence of the eyes in the child’s head. Craniofacial proportions change with the onset of puberty. The midface and lower face elongate and protrude, producing the definitive adult face with its sexual dimorphism. Aging from infancy to adulthood is, therefore, associated not only with enlargement of the head, but also changes in shape and proportions of its components; this is called allometric growth.
Although the cellular dynamics of the progression from zygote to embryonic germ layers are poorly understood, the importance of interplay among neurectoderm, surface ectoderm, mesoderm, and endoderm in the development of the head is appreciated. Cephalic neural crest is believed to be the source of most craniofacial mesenchyme, a process that requires migration and loss of epithelial differentiation. Early craniofacial embryonic development is characterized by the formation of headfolds, which are outgrowths that form and surround cavities or grooves, which then close either totally or partially by the
continued enlargement of the outgrowths. In addition, surface ectoderm placodes develop, which contribute to the eyes (optic placode), nose (nasal placode), ear (acoustic placode), and branchial arches (epibranchial placode). Outpouching of the ventral foregut creates pharyngeal pouches that interact with overlying surface ectoderm and mesenchymal cells at the site of the developing branchial grooves.
continued enlargement of the outgrowths. In addition, surface ectoderm placodes develop, which contribute to the eyes (optic placode), nose (nasal placode), ear (acoustic placode), and branchial arches (epibranchial placode). Outpouching of the ventral foregut creates pharyngeal pouches that interact with overlying surface ectoderm and mesenchymal cells at the site of the developing branchial grooves.
As the adjacent branchial arches enlarge, the grooves deepen. Five branchial arches, separated by four grooves, arise in the human embryo. The middle and lower face arise from these arches, with the first arch being dominant. The central portion of the face arises from the frontonasal process and then fuses with the paired maxillary processes of the first branchial arches. Where facial swellings contact each other, an epithelial plate develops, which either degenerates or persists as an epithelial lines structure, for example, the nasolacrimal duct, depending on location. Facial muscles develop by interaction between specific nerves and focal mesenchyme. The craniofacial skeleton arises from a number of discrete ossification centers with one or, in a few cases, two centers per definitive named bone. Two types of bone develop in the head—membranous and endochondral. Membranous bone comprises most of the cranial vault and facial bones; endochondral bones comprise most of the cranial base.
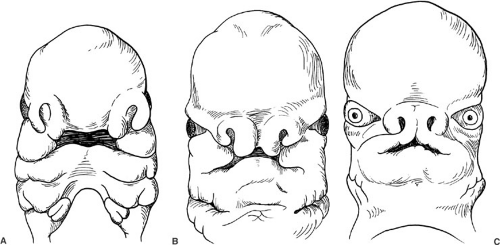 FIGURE 53-1. (A) At 5 weeks’ gestation, the human embryo has developed grooves between the central and lateral portions of the face. Figure 53-10 depicts a child in whom a midline cleft suggests the right and left medial elements have not completely fused. (B) At 6 and one-half weeks, the embryo has widely spaced eyes. When orbital migration toward the midline does not subsequently occur, ocular hypertelorism results, as in Fig. 53-4. (C) By 7 weeks’ gestation, the primordial external ear begins to form in the neck. An absence of the external ear and auditory meatus, as depicted in Fig. 53-11, suggests either a basic regional anatomic error in the genome or disruption of a cellular differentiation/migration process. |
Current Concepts Regarding Abnormal Development
There are two types of craniofacial anomalies—malformations and deformations. Malformation means that the part never formed correctly; deformation means that the part formed correctly, but was secondarily deformed. Neither term indicates the process whereby the malformation or deformation occurred. Some craniofacial anomalies are hereditary and, in an increasing number of cases, the abnormal chromosome has been identified. Some result from recognized teratogens, and some are believed to be the result of intrauterine constraint, vascular accidents, or amniotic bands. Most are sporadic and their induction factors unknown (1,5).
Disorders of cell proliferation, degeneration, differentiation, and migration, as well as in the composition of the extracellular matrix, have been postulated as the mechanism whereby malformations occur. Although there is no consensus on classification of the etiologies of craniofacial anomalies or their dysmorphology, several useful schemes have been proposed. Based on current concepts of embryology, Vermeij-Keers (6) proposed three different groups of malformations: cerebrocranial dysplasia (malformations of the brain and cranium), cerebrocraniofacial dysplasia (facial defects involving the brain or eyes and the cranium), and craniofacial dysplasias (defects of the face
and cranium alone). These groupings help understanding of specific anomalies in light of current concepts of embryology. In contrast, Tessier (7) proposed a topographic soft- and hard-tissue map for classifying craniofacial anomalies, which has been widely adopted to facilitate communication about rare defects.
and cranium alone). These groupings help understanding of specific anomalies in light of current concepts of embryology. In contrast, Tessier (7) proposed a topographic soft- and hard-tissue map for classifying craniofacial anomalies, which has been widely adopted to facilitate communication about rare defects.
The dysmorphology of some craniofacial anomalies seems to document arrest of embryogenesis (Fig. 53-1A). Others seem to portray failures of fusion (Fig. 53-1B), and in others, a gross disruption of spatial processes (Fig. 53-1C). The similarity between an anomaly and an embryonic stage, however, does not necessarily mean that the two are directly related. Current research on homeobox genes (4), the “blueprint” for anatomic organization, and the biochemical aspects of genetic expression should yield additional understanding of the origin and expression of craniofacial anomalies.
THE CRANIOFACIAL DEFORMITIES TEAM: INTERDISCIPLINARY CARE
Craniofacial anomalies affect both the skull and the face; however, some major facial anomalies without cranial abnormalities are also categorized as craniofacial anomalies because of the extent of involvement. No single health care discipline contains all the expertise necessary for evaluating, treating, and following a patient with a craniofacial anomaly. For this reason, the standard for contemporary care of craniofacial anomalies involves a craniofacial interdisciplinary team. The craniofacial deformities team developed from the cleft lip and palate team by adding the neurosciences and ancillary services necessary to execute complex operations. The team approach has been endorsed by a consensus conference on clefts and other craniofacial anomalies conducted by the American Cleft Palate/Craniofacial Association and funded by the U.S. Department of Maternal and Child Health (8). A directory of craniofacial teams is maintained by the American Cleft Palate/Craniofacial Association, 1504 East Franklin Street, Suite 102, Chapel Hill, NC 27514, and can be obtained by phoning 1-800-24 CLEFT (http://www.cleftline.org).
The disciplines represented by the craniofacial team usually include audiology, genetics, neurosurgery, nursing, ophthalmology, orthodontics, oral-maxillofacial surgery, otolaryngology, pediatric dentistry, pediatrics, plastic/craniofacial surgery, prosthodontics, psychology/psychiatry, social services, and speech/language pathology (9). Anesthesiology, computer services, neurology, and radiology are important components of surgical planning and execution, but usually do not participate as regular team members. Full craniofacial team evaluations are often conducted on an annual basis from 1 to 4 years of age, every 2 years from ages 4 to 14 years, and then at ages 17 and 20 years, when the patients are then graduated from the program. Patients with new or persistent problems are seen more frequently by whichever providers are appropriate.
The primary purposes of the craniofacial team are to formulate and update a long-term care plan, to coordinate recommendations with primary and secondary local providers, and to provide tertiary services. Although details of craniofacial team function vary from center to center, standard elements include
Regularly scheduled ambulatory multidisciplinary patient evaluations
Collation of individual provider recommendations into a coherent, efficient, comprehensive care plan
Production of a written report for the patient’s primary and secondary care providers
Production of a written interpretive letter for the parents and patient, if of appropriate age
Monitoring of status of treatment recommendations
A program of quality assurance
Maintenance of permanent records with standard documentation at regular intervals using text, photographs, and diagnostic medical imaging
DIAGNOSTIC AND OUTCOME CRANIOFACIAL EVALUATION
Diagnosis begins with a complete history and physical examination. This evaluation should include the parents and, when possible, any family members who are suspected of having similar disorders. If a positive family history for craniofacial deformities is elicited, referral to a medical geneticist versed in dysmorphology is indicated.
Craniofacial clefts, aplasias, and severe hypoplasias are usually recognized at birth. Abnormalities of head shape and mild-to-moderate hypoplasias may initially be interpreted as the result of the birthing process and not appreciated as anomalies until several months after birth.
A complete physical examination of the head and neck should be performed, regardless of how focal the anomaly seems because many seemingly isolated anomalies have subtle associated findings. The general appearance of the patient with respect to alertness, ease of respiration, age-appropriateness of behavior, and nutritional status should be noted. The head is assessed for symmetry and proportion. Function of cranial nerves II through VIII and XII is discretely tested as are the combined functions of IX through XI for swallowing and speech. The scalp is inspected for hair pattern, alopecia, and masses. With respect to the cranium, perimeter shape, status of the fontanelles, osseous integrity, presence or absence of sutural ridges, and forehead, supraorbital, and nasofrontal configurations are recorded. The eyes are examined for the relation between the globes and the orbits, the distance between the pupils in central conjugate gaze, the medial
canthal distance, the palpebral fissure orientation, eyelid continuity and function, eyelash orientation, lacrimal function, globe motion, and visual status. Examining the midface, the physician should note the orientation of the nasal dorsum and its height, the configuration of the nasal tip and its projection, the placement of the nasal septum, the status of the intranasal mucosa, the patency of each nasal airway, and the soft and hard tissues of the cheeks. The ears, including the external auditory meati, are evaluated for placement, shape, and size. The lips are inspected for clefts, cysts, or pits, and the labial competency at rest and during speech and eating is evaluated. The intraoral examination includes temporomandibular joint function, the physical and occlusal dental status, the dentoskeletal maxillary–mandibular relationship, lingual and palatal anatomic integrity and function, and the oropharyngeal lymphoid tissues. The range of motion of the neck is tested, spontaneously and provocatively, and the neck is palpated for restrictive muscles and masses. An experienced craniofacial examiner can conduct this evaluation in a brief period and then focus on the pathology detected.
canthal distance, the palpebral fissure orientation, eyelid continuity and function, eyelash orientation, lacrimal function, globe motion, and visual status. Examining the midface, the physician should note the orientation of the nasal dorsum and its height, the configuration of the nasal tip and its projection, the placement of the nasal septum, the status of the intranasal mucosa, the patency of each nasal airway, and the soft and hard tissues of the cheeks. The ears, including the external auditory meati, are evaluated for placement, shape, and size. The lips are inspected for clefts, cysts, or pits, and the labial competency at rest and during speech and eating is evaluated. The intraoral examination includes temporomandibular joint function, the physical and occlusal dental status, the dentoskeletal maxillary–mandibular relationship, lingual and palatal anatomic integrity and function, and the oropharyngeal lymphoid tissues. The range of motion of the neck is tested, spontaneously and provocatively, and the neck is palpated for restrictive muscles and masses. An experienced craniofacial examiner can conduct this evaluation in a brief period and then focus on the pathology detected.
Diagnostic medical imaging is an important part of evaluating a patient with a craniofacial anomaly. Increasingly, craniofacial anomalies are being identified prenatally with ultrasonography. Counseling is provided to the parents prenatally and perinatally in such cases. After birth, routine skull X-rays are useful for identifying premature fusion of cranial sutures (craniosynostosis), ultrasonography for identifying abnormal fluid collections (e.g., hydrocephalus or cystic hygroma), computed tomography (CT) scans for osseous anomalies, and magnetic resonance (MR) scans for intracranial and extracranial soft-tissue anomalies, including vascular malformations. The development of software to produce surface-shaded and volumetric three-dimensional skeletal and soft-tissue images from CT or MR digital data has greatly increased our knowledge of craniofacial anomalies and our ability to plan complex operations, assess the execution of surgical plans, and evaluate the outcome longitudinally (10,11).
SURGICAL MANAGEMENT OF CRANIOFACIAL ANOMALIES
Intracranial and Extracranial Procedures
Cranium
The two major categories of cranial anomalies that demand the attention of the craniofacial surgeon are abnormal shape and masses. Deformation of cranial shape is a common consequence of birth and should correct to a normal shape within the first few weeks of life. Persistence of an abnormal cranial shape beyond the second month of life is cause for evaluation and management as appropriate. Although a cranial mass noted perinatally may be an innocent cephalohematoma that will resolve over time, those that persist need to be evaluated for intracranial extension.
Premature fusion of cranial sutures, craniosynostosis, causes abnormal cranial shape due to a combination of growth restraint, secondary to the synostosis, and compensatory deformation, secondary to pressure exerted by the growing brain on those bones not restricted by synostosis (Fig. 53-2). In addition to the cranial abnormality, craniosynostosis may produce deformity of the orbit, midface, or lower face. Surgical intervention has sought to ablate the synostotic suture. It was believed that craniosynostosis produced increased intracranial pressure, hydrocephalus, blindness, and mental retardation. These dire prognostications are excessive. Few patients with craniosynostosis have such associated morbidity, and most who do are those with syndromic synostosis involving multiple sutures. However, some cognitive and/or behavioral impairment is associated with even the nonsyndromic craniosynostoses. In some cases of single-suture nonsyndromic craniosynostosis and in many cases of multiple-suture synostosis, low-grade increased intracranial pressure is noted (12). The neurologic significance of this finding remains a subject of debate. Operative correction of calvarial and superior orbital deformities, and an attempt to prevent subsequent secondary facial deformation, are the surgical goals in treating craniosynostosis. Collaboration between craniofacial surgeons and neurosurgeons has led to better outcomes more recently.
The relation between abnormal head shape and restriction of neck mobility is of interest to craniofacial, pediatric, and orthopedic surgeons, as well as pediatricians and physical therapists. It has long been recognized that limited head/neck motion in an infant can result in cranial deformity, such as occipital flattening (brachycephaly) secondary to papoose board swaddling, anteroposterior elongation and bitemporal narrowing (scaphocephaly) secondary to prematurity, and asymmetry (plagiocephaly) secondary to torticollis. Cranial deformity with limited neck motion is not always a case in which the cranial problem follows the cervical one, however. A common type of asymmetric skull, known as positional or deformational plagiocephaly, usually develops postnatally due to sleep position in an otherwise normal infant, and the infant’s persistence in lying on the flattened side of the head can produce a secondary neck dysfunction due to tightening of the ipsilateral neck muscles and lengthening of those contralateral. If the process is not interrupted with physical therapy or correction of the plagiocephaly, the neck dysfunction can progress to a fixed limitation in range of motion that in turn aggravates the plagiocephaly. Although physical therapy can improve the neck range of motion, it does not correct the skull deformity. Positional or deformational plagiocephaly can be identified by the history, physical examination, and documentation of radiolucent sutures on screening skull X-rays. For patients younger
than 12 months, plagiocephaly without synostosis is managed with cranial molding helmets (Fig. 53-3) (13). After 1 year of age, the child’s skull is too hard to respond, and the only option is surgical cranial reconstruction.
than 12 months, plagiocephaly without synostosis is managed with cranial molding helmets (Fig. 53-3) (13). After 1 year of age, the child’s skull is too hard to respond, and the only option is surgical cranial reconstruction.
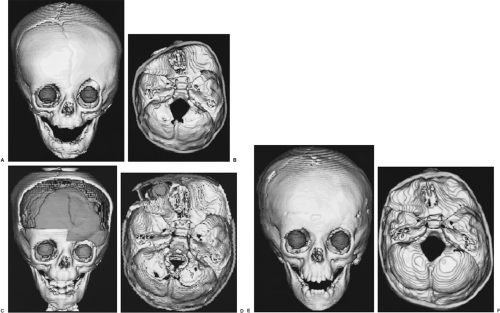 FIGURE 53-2. Three-dimensional CT osseous reformations with globe opacification of a child with craniosynostosis that caused abnormal cranial shape. (A) Preoperatively, there is a ridge where the left coronal suture should be, the orbital rims are asymmetric, and the left orbit is vertically elongated. (B) The view of the endocranial base preoperatively shows that the anterior hemicranial fossa ipsilateral is compressed against the synostosis and that the contralateral has expanded, “unroofing” the ipsilateral globe. (C) Perioperatively, the reshaped and repositioned frontal bones and right superolateral orbit appear gray. The brain is seen through the bicoronal craniectomy defect. (D) The basal view perioperatively shows that the right frontal bone and superolateral orbital rim (gray) have been moved ventrally and the left frontal bone moved dorsally to achieve anterior fossa symmetry and appropriate orbital roof depth. The globe (gray sphere) is visible through the ventral enlargement of the orbital roof. The placement of an autologous calvarial graft (strut lateral to the globe) maintains the orbital rim advancement. (E) One year postoperatively, the skull has reossified while maintaining the perioperative symmetry of the orbits and without regenerating a left coronal suture. (F) The symmetry of the normalized anterior fossa has been maintained. The asymmetric posterior fossa remains the same as seen preoperatively. (See Color Fig. 53-2C and D.)
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|