Fetal Breathing
Studies in animal models and human fetuses have demonstrated a maturational progression in fetal breathing movements during gestation that can be altered by a variety of pharmacologic and physiologic inputs. In humans, fetal breathing movements have been characterized in response to maternal condition and as a potential indicator of overall fetal condition. Regardless of gestational age, fetal breathing is not a continuous process; significant periods of apnea lasting as long as 2 hours occur even in near-term fetuses and are generally more frequent and of longer duration at younger gestational ages (
1). During periods of frequent respiratory motion, patterns of both regular and irregular breathing are documented by chest/abdominal wall movements and by Doppler sonography assessing tracheal fluid flow (
2). Fetal breathing movements exhibit a circadian rhythmicity, with well-documented increases during certain periods of the day. Maternal condition, particularly maternal glucose status, can have significant effects on fetal breathing frequency, with well-documented increases in fetal breathing frequency after maternal glucose loading. This response to maternal glucose loading is most pronounced when the mother is fasting (
3).
Fetal breathing has some clinical utility when assessing fetal well-being. Several studies have documented diminished fetal breathing activity in association with poor fetal health, and this decreased activity, along with other measures of fetal well-being, can be helpful in guiding obstetric management (
4). The fetal breathing pattern responds to a variety of pharmacologic and physiologic manipulations. Fetal breathing increases if the mother inhales CO
2 (
5). Maternal hyperoxia does not alter fetal breathing movements or pattern in near-term normally grown fetuses, but growth-restricted fetuses exhibit an increase in respiratory rate with maternal hyperoxia (
6). Tocolytics, indomethacin, and terbutaline increase fetal breathing movements when administered during preterm labor (
7).
Animal studies augment the human ultrasound data and provide a clearer picture of the maturational development of breathing control. Fetal breathing activity in sheep starts early in gestation, arises from centrally mediated stimuli, and occurs primarily during periods of low-voltage electrocortical activity (REM sleep) (
8). REM sleep comprises about 40% of fetal life during the last trimester in sheep. Breathing also occurs during periods of high-voltage electrocortical activity (quiet sleep), but it is only episodic and generally associated with muscular discharges (
9). Animal data have also confirmed that the fetal breathing pattern changes in response to physiologic derangements (i.e., hypercarbia, hypoxia, hyperoxia) (
10,
11). In response to hypercapnia, the fetus increases respiratory rate and tidal volume, suggesting intact central chemoreception. The fetal response to hypoxia appears to be centrally mediated and results in diminished neuronal activity and diminished or absent fetal breathing movements, but the peripheral chemoreceptors may also contribute to absence of the “adult” response to hypoxia of an increase in respiratory rate and tidal volume.
Initiation of Breathing at Birth
Despite the enhanced understanding of fetal maturation of breathing control, our understanding remains incomplete regarding factors responsible for initiating and maintaining a regular pattern at birth (
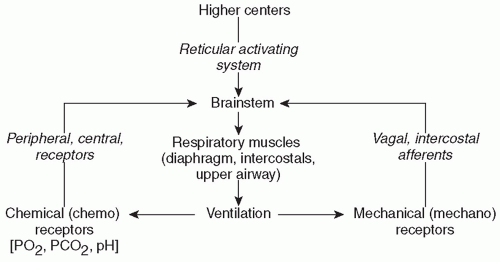
12). Development and maintenance of respiration in the newborn are likely due to a complex interaction of sensory stimuli and both central and peripheral chemoreceptor inputs (
Fig. 25.1). The basal level of fetal chemoreceptor discharge adapts to the fetal PaO
2, and the several-fold increase in PaO
2 at birth silences the chemoreceptors (
13). During the fetal-to-neonatal transition, however, peripheral chemoreceptors may not be completely silenced, as evidenced by the fact that supplemental O
2 compared to room air at birth may delay onset of the first cry and of sustained ventilation thereafter. The degree of maturation in the central respiratory centers also appears to be important since the responses to respiratory stimuli in the term infant are more developed than in the preterm infant.
Neonatal Breathing
Studies in human and animal neonates have provided important insights into the understanding of respiratory rhythm generation (
14). The control and maintenance of normal breathing largely reside within the respiratory control centers of the bulbopontine
region of the brainstem. Neurons within this area respond to multiple afferent inputs to modulate their own inherent rhythmicity and provide efferent output to the respiratory control muscles. Multiple afferent inputs induce modulation of the central respiratory center efferent outputs to the lungs and respiratory and airway muscles. These afferent inputs are “categorized” by the respiratory control center; some inputs cause an instantaneous response in the control center output, while others only act to “shape” the respiratory response, resulting in small changes in muscular output, tidal volume, and airway tone (
15). Among these inputs are signals from central and peripheral chemoreceptors, pulmonary stretch receptors, and cortical and reticuloactivating system neurons. These afferent inputs and resultant efferent outputs of the central respiratory center are summarized in
Figure 25.1. Sleep state can also have a profound effect on respiratory pattern, although sleep state is often difficult to classify in preterm infants.
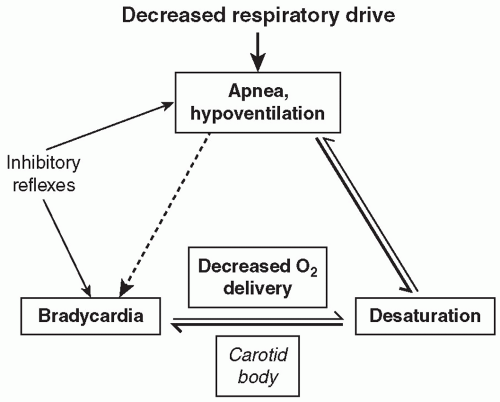
In the adult, these multiple afferent inputs act upon the neurons within the respiratory control center and provide a well-integrated response to perturbations in the system and characteristic respiratory patterns. For example, increased respiratory rate and depth will characteristically result from activation of central chemoreceptors in response to hypercarbia. In the newborn infant and especially in the preterm infant, however, these responses are less well organized and of lesser magnitude, and apnea with associated desaturation and/or bradycardia is a common result of this disorganized or immature response to multiple afferent inputs.
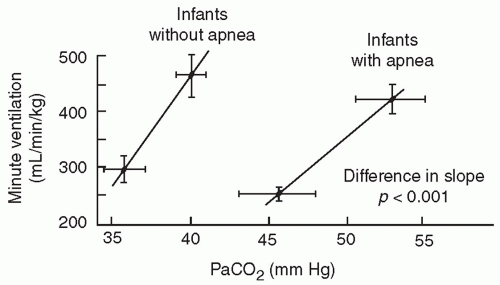
The central chemosensitivity to hypercarbia is diminished in infants born preterm and is unrelated to any mechanical limitations of ventilation (
16). The slope of the CO
2 response curve (
Fig. 25.2) increases significantly between 29 to 32 and 33 to 36 weeks of gestation, especially in infants without clinically evident apnea. The cause of this decreased sensitivity of central respiratory centers to CO
2 in preterm infants is related to central nervous system immaturity as indicated by decreased synaptic connections and incomplete dendritic arborization.
Preterm infants and full-term infants up to approximately 3 weeks’ postnatal age have a characteristic biphasic response to hypoxia that is quite different than in older infants. In contrast to the sustained hyperventilation in older infants, preterm infants have only a transient hyperventilation lasting 30 seconds to a minute, followed by progressive ventilatory depression despite continued low inspired oxygen concentrations. The initial hyperventilatory response may be completely blunted in extremely premature infants. This initial transient hyperventilation is likely in response to peripheral chemoreceptor input, and the subsequent hypoxic depression appears to be at least primarily due to centrally mediated decreased peripheral chemoreceptor activity secondary to descending inhibition from the upper brainstem, midbrain, or higher structures (
16).
In addition to maturation of central chemoreception, peripheral chemoreceptors progressively mature over the first weeks of life in both full-term and preterm infants, as manifested by the decrease in ventilation with hyperoxia and the hypoxic ventilatory response (
13). The magnitude of the ventilatory depression occurring with acute hyperoxia is relatively blunted in infants born preterm, and the magnitude of the hypoxic ventilatory depression is greater in preterm infants with symptomatic AOP. However, it is still unclear to what extent, if any, immature or mature peripheral chemoreception is associated with apnea symptoms. Peripheral chemoreceptor activation plays a role in apnea termination, but excessive peripheral chemoreceptor activation may destabilize the respiratory pattern in AOP and exacerbate the extent of apnea and associated bradycardia and intermittent hypoxia. Carotid chemoreceptors may thus be important not only in stimulating arousal from apnea-associated desaturation, and hence apnea termination, but also potentiating the risk of occurrence. The extent of apnea does seem to correlate with maturation of the ventilatory response to acute hypoxia, with less apnea and intermittent hypoxia present in the early weeks of life and increased apnea-related symptoms in the later weeks with maturation of carotid chemoreceptivity and greater sensitivity to acute hypoxia (
13).
Both central and peripheral inputs are thus likely important in the onset and termination of apnea (
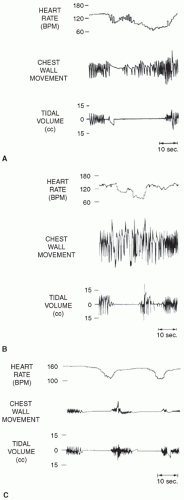
16). The coordination of phasic and tonic inputs to determine breathing rhythms is not yet fully understood, but stable autonomic control of breathing appears to involve multiple phasic, random, and descending inputs. Upper airway reflexes also may play a role in inhibiting respiration, particularly in preterm infants. Multiple sensory afferent fibers exist within the upper airways, and stimulation of these fibers by various mechanisms can result in abnormal respiratory responses. Responses to upper airway afferent fiber stimulation can change markedly with maturation. Negative pressure in the upper airways in human infants results in depressed ventilation. This inhibition may contribute to the central apnea that often follows obstructed breaths (
Fig. 25.3). As upper airway obstruction occurs, the infant makes respiratory efforts against this obstruction, and the resulting increased negative pressure in the upper airway may result in reflex inhibition of diaphragmatic contraction. Due to a blunted response to hypercarbia and hypoxic ventilatory depression, less mature preterm infants with apnea may be unable to recover spontaneously and hence be more likely to require active intervention.
Activation of laryngeal mucosal receptors can elicit strong airway protective reflexes in both full-term and preterm infants. This laryngeal chemoreflex can result in autonomic responses including apnea, bradycardia, hypotension, upper airway closure, and swallowing (
16). Although this chemoreflex is an important contributor to aspiration-related apnea and bradycardia, there is no clear relationship to AOP.
Genetics of Control of Breathing
Recent genetic studies related to brainstem autonomic regulation have enhanced our understanding of normal development of respiratory regulation (
17). Targeted gene inactivation studies in animals have identified multiple genes involved with prenatal brainstem development of respiratory control including arousal responsiveness. During embryogenesis, the survival of specific cellular populations composing the respiratory neuronal network is regulated by neurotrophins, a multigene family of growth factors and receptors. Brain-derived neurotrophic factor (BDNF) is required for development of normal breathing behavior in mice, and newborn mice lacking functional BDNF exhibit ventilatory depression associated with apparent loss of peripheral chemoafferent input. Ventilation is depressed, and hypoxic ventilatory drive is deficient or absent.
Krox-20, a homeobox gene, appears to be required for normal development of the respiratory central pattern generator (
18).
Krox-20 null mutants exhibit an abnormally slow respiratory rhythm and increased incidence of respiratory pauses, and this respiratory depression can be further modulated by endogenous enkephalins. Absence of
Krox-20 may result in the absence of a rhythm-promoting reticular neuron group localized in the caudal pons and could thus be a cause of life-threatening apnea.
Brainstem muscarinic cholinergic pathways are important in ventilatory responsiveness to carbon dioxide (CO
2). The muscarinic system develops from the neural crest, and the
ret protooncogene is important for this development (
17).
Ret knockout mice have a depressed ventilatory response to hypercarbia, implicating absence of the
ret gene as a cause of impaired hypercarbic responsiveness. Diminished ventilatory responsiveness to hypercarbia has also been demonstrated in male newborn mice heterozygous for
Mash-1. There is a molecular link between
ret and
Mash-1, and the latter is expressed in embryonic neurons in vagal neural crest derivatives and in brainstem locus coeruleus neurons, an area involved with arousal responsiveness.
Serotonin (5-HT) is a widespread neurotransmitter that affects cardiovascular control and modulates activity of the circadian clock. Serotonergic receptors in the brainstem are critical components of respiratory drive. Multiple genes are involved in the control of serotonin synthesis, storage, membrane uptake, and metabolism (
17). Polymorphisms have been identified in the promoter region of the 5-HT transporter protein gene located on chromosome 17, and variations in the promoter region of the gene appear to have a role in serotonin membrane uptake and regulation. Several transporter polymorphisms have been described that may occur in greater frequency in SIDS than in control infants, but no data are available related to maturation of breathing control in preterm infants in general or to AOP in particular. Thus, there are no data on the potential role of serotonin-related polymorphisms in determining the extent of clinical manifestations of AOP. However, the greater concordance for AOP among monozygotic twins than same-sex dizygotic twins suggests a genetic contribution (
19).
These studies illustrate potentially important genetic foundations of neonatal control of breathing. Further work is needed, however, to better understand the developmental regulation of these targeted genes and their influence on maturation of the fetal/neonatal respiratory centers and peripheral chemoreceptors.