Congenital Heart Disease
Stuart Berger
James S. Tweddell
Medical College of Wisconsin, Children’s Hospital of Wisconsin, Milwaukee, Wisconsin 53226.
Medical College of Wisconsin, Children’s Hospital of Wisconsin, Milwaukee, Wisconsin 53226.
Five to 8 out of 1,000 live born infants are diagnosed with a congenital cardiovascular defect (1,2,3) making congenital heart disease the most common birth defect. Cardiovascular abnormalities continue to be the leading cause of premature mortality from congenital abnormalities. The spectrum of cardiovascular involvement is quite variable and may include defects with minimal physiologic impact, such as a small ventricular septal defect, or more complex lesions, such as tetralogy of Fallot, which requires surgical intervention with a lifelong risk of sequelae.
Using data from the Pediatric Health Information System, an administrative database maintained by 32 major pediatric hospitals in the United States, the mean hospital charge for a patient undergoing surgical intervention for congenital heart disease is $88,042 per patient from 1997 to 2000 (4). The American Heart Association estimates that 40,000 children are born with congenital heart disease each year (5). In addition, there are 1,000,000 Americans with congenital heart disease alive today. If we assume that 25% of patients with congenital heart disease require hospital intervention for their congenital heart defect during the first year of life, and that 5% of the remaining patients with congenital heart disease older than 1 year will require a procedure each year, then the hospital charges for the diagnosis of congenital heart disease will be more than $5 billion per year. It should be noted that this figure underestimates the economic impact of congenital heart disease because it does not take into account all physician charges, outpatient medication costs, home health care costs, or days of school and work lost by patients and family members. It is clear that the cost and impact of congenital heart disease for the individual, the family, and society is large.
ETIOLOGY OF CONGENITAL HEART DISEASE
As much as 6% of congenital heart disease is associated with genetic factors, whereas 2% are known to be related to environmental causes. Well-known examples of environmental causes of congenital heart disease include rubella virus, which is associated with patent ductus arteriosus and branch pulmonary artery stenosis, and thalidomide, which is associated with tetralogy of Fallot and atrial septal defects. Additional environmental causes of congenital heart disease were identified by the Baltimore-Washington Infant Study (BWIS), a regional epidemiologic study of congenital heart disease (3,6,7). The BWIS took on the difficult task of identifying congenital heart disease among live births in a contiguous region of Maryland and northern Virginia from 1981 to 1986. Epidemiologic techniques were used to link congenital heart defects with environmental and genetic factors. Environmental factors apparently associated with congenital heart disease included paternal exposure to cold temperature and maternal exposure to solvents and hair dyes. In addition, the risks were increased with maternal exposure to diazepam, corticosteroids, phenothiazine, and gastrointestinal drugs. Paternal exposure to cocaine was also implicated. Other associations included an apparent link between maternal diabetes and tetralogy of Fallot, truncus arteriosus, and double-outlet right ventricle. Table 62-1 lists the reported associations of environmental influences and congenital heart disease.
Heritable syndromes, chromosomal abnormalities, and increasingly, specific gene defects have been linked to congenital cardiac abnormalities (8). Table 62-2 lists the syndromes and associated congenital cardiac defects, along with specific gene associations and/or chromosomal abnormalities where they are known. Mutation of the gene FBN1 that codes for fibrillin, a component of the aortic wall, results in Marfan syndrome (9). This disorder demonstrates autosomal dominant expression, and all
heterozygotes express the trait to some degree. In addition to Marfan syndrome, trisomy 18 is associated with congenital heart disease in 100% of cases (10). In trisomy 18, however, the specific form of congenital heart disease is variable. Although ventricular septal defects predominate, other forms of congenital heart disease can occur. In other well-defined genetic syndromes, the association with congenital heart disease is less constant. For example, only 50% of children with Down syndrome (trisomy 21) have a congenital cardiac abnormality. The variable expression of congenital heart disease with a definite chromosomal abnormality is also demonstrated among patients with 22q11 deletion, where about one-half the affected individuals will have a conotruncal defect, an abnormality of the outflow of the heart and great vessels (11). Examples of specific gene mutations resulting in congenital heart disease include the gene NKX2-5, on chromosome 5, that results in atrial septal defects and conduction abnormalities (12). Holt-Oram syndrome, characterized by limb abnormalities and defects of the atrial septum, is caused by a mutation in the TBX5 gene located on chromosome 12 (13). The association of limb malformations and cardiac defects, “heart-hand syndromes,” with a single gene defect suggests that the same gene may be responsible for several developmental processes. The more recent identification of several ion channel gene defects (HERG, SCN5A, KVLQT1, and MinK) associated with long QT syndrome have confirmed the genetic basis of this group of arrhythmias, which are known to be familial (14). The identification of genes fundamental for development of left–right asymmetry in animal models, such as nodal and Lefty2, defects of which are associated with cardiac abnormalities similar to heterotaxy syndromes in humans, suggests the role of homologous genes in congenital heart disease in humans (15). Genes responsible for cardiac development are being increasingly identified and mapped. Corresponding mutations that result in heart defects are being identified. Researchers will undoubtedly begin to identify additional gene mutations that result in isolated congenital heart defects. Although presently the cause of 85% of congenital heart disease is unknown, it is likely due to a combination
or interaction between environmental causes and genetic susceptibility.
heterozygotes express the trait to some degree. In addition to Marfan syndrome, trisomy 18 is associated with congenital heart disease in 100% of cases (10). In trisomy 18, however, the specific form of congenital heart disease is variable. Although ventricular septal defects predominate, other forms of congenital heart disease can occur. In other well-defined genetic syndromes, the association with congenital heart disease is less constant. For example, only 50% of children with Down syndrome (trisomy 21) have a congenital cardiac abnormality. The variable expression of congenital heart disease with a definite chromosomal abnormality is also demonstrated among patients with 22q11 deletion, where about one-half the affected individuals will have a conotruncal defect, an abnormality of the outflow of the heart and great vessels (11). Examples of specific gene mutations resulting in congenital heart disease include the gene NKX2-5, on chromosome 5, that results in atrial septal defects and conduction abnormalities (12). Holt-Oram syndrome, characterized by limb abnormalities and defects of the atrial septum, is caused by a mutation in the TBX5 gene located on chromosome 12 (13). The association of limb malformations and cardiac defects, “heart-hand syndromes,” with a single gene defect suggests that the same gene may be responsible for several developmental processes. The more recent identification of several ion channel gene defects (HERG, SCN5A, KVLQT1, and MinK) associated with long QT syndrome have confirmed the genetic basis of this group of arrhythmias, which are known to be familial (14). The identification of genes fundamental for development of left–right asymmetry in animal models, such as nodal and Lefty2, defects of which are associated with cardiac abnormalities similar to heterotaxy syndromes in humans, suggests the role of homologous genes in congenital heart disease in humans (15). Genes responsible for cardiac development are being increasingly identified and mapped. Corresponding mutations that result in heart defects are being identified. Researchers will undoubtedly begin to identify additional gene mutations that result in isolated congenital heart defects. Although presently the cause of 85% of congenital heart disease is unknown, it is likely due to a combination
or interaction between environmental causes and genetic susceptibility.
TABLE 62-1 Associations of Environmental Influences and Congenital Heart Disease. | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||
TABLE 62-2 Syndromes and Associated Congenital Cardiac Defects Along with Specific Gene: Associations and Chromosome Locations. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
CARDIAC DEVELOPMENT
Congenital heart disease is by definition present at birth and is due to abnormalities of development. Most forms of congenital heart disease appear to be the result of arrested development, and therefore an understanding of basic cardiac development is essential to understanding congenital heart disease and its treatment. The vascular system of the human embryo begins to form when the developing embryo is no longer able to sustain nutritional requirements by diffusion alone; this occurs during the third gestational week (16). Two endocardial heart tubes develop at 18 to 20 days of gestation at the cephalad end of the embryo fusing into a single heart tube at 22 days of development. From caudad to cephalad, the heart tube is divided into segments: (1) sinus venosus, (2) primitive atria, (3) ventricle, and (4) bulbus cordis. These four segments will respectively develop into (1) the right atrium and coronary sinus, (2) the left atrium (3) the left ventricle, and (4) the right ventricle and outflow tracts of both ventricles. Differential growth of heart tube compared with the remaining embryo cause it to bulge ventrally (anteriorly) into the surrounding pericardial sac, and differential growth of the heart tube itself results in a rightward looping of the developing heart. By 28 days of development, the bulboventricular loop has formed and septation of the atrium has begun. The division of the ventricles and atrioventricular (AV) valves is initiated by the growth of the interventricular septum from the floor of the common ventricle at the bulboventricular sulcus (Fig. 62-1), and by the endocardial cushions that originate at the junction of the primitive atrium and left ventricle. Initially, this common AV orifice provides access only to the left ventricle; blood can only enter the primitive right ventricle (derivative of the bulbus cordis) through the bulboventricular foramen. With ongoing growth, the AV canal enlarges to the right (Fig. 62-1b). This provides direct access of the right side of the common AV valve, destined to become the tricuspid valve, to the bulbus cordis, which is destined to become the right ventricle. The endocardial cushion separates the common AV valve into the tricuspid and mitral valves. The valves themselves form from tissue at the AV junction, and the subvalvar apparatus forms from muscular cords that form from the endocardial surface and eventually develop into the chordae tendineae and papillary muscles (Fig. 62-2). Ventricular septation is completed by the seventh week. Separation of the outflow into pulmonary and systemic circuits is initiated during the fifth week of gestation by swellings on the right and left sides of the conus, and by right superior and left inferior swellings on the truncus arteriosus (Figs. 62-1b and c). Fusion of these opposing swellings results in the formation of the aorticopulmonary septum dividing the truncus into an aortic and pulmonary channel. The swellings within the conus separate the right and left ventricular outflow tracts and terminate proximally to complete ventricular septation. Formation of the heart is completed by 49 days of gestation. Congenital heart disease is nearly always the result of arrest of a portion of cardiac development. Common problems include defects of septation, such as atrial septal defects, ventricular septal defects, and AV septal defects, which result from failure of any of the components of the developing atrial and ventricular septa; conotruncal truncal abnormalities that result from failure of the outflow and proximal great vessels to complete septation and achieve proper alignment; and finally, lesions involving underdevelopment of the inflow or outflow of either ventricle that will result in a spectrum of hypoplastic lesions of either the left or right heart.
FETAL CIRCULATION
The demands of a permanent terrestrial existence required the development of a dual circulatory system: a low-pressure system for pumping desaturated blood through a low-resistance pulmonary circuit and a high-pressure system for pumping the oxygenated blood to the tissues. Although cardiac development is completed during the seventh week of development and results in two circulatory systems within one heart, the developing human is still more than 7 months away from breathing air. A functional single ventricle or Piscean circulatory system is maintained by the fetal circulatory pattern, which allows both the right and left sides of the heart to contribute to systemic work prior to delivery (16). Figure 62-3 is a drawing of the fetal circulation. In the fetus, gas exchange takes place in the placenta. Blood returning from the placenta via the single umbilical vein enters the fetus through the umbilicus and then empties into the inferior vena cava (IVC) via the ductus venosus. The IVC blood, with the umbilical vein contribution and therefore slightly higher oxygen saturation than blood from the superior vena cava (SVC), enters the right atrium and is preferentially shunted to the left atrium through the foramen ovale. The left ventricle now pumps this more oxygen-rich blood to the cerebral circulation. The right ventricle also performs systemic work by pumping blood to the descending aorta via the ductus arteriosus. The umbilical arteries arising from the iliac arteries now return to the placenta for gas and nutrient exchange.
Profound changes in the circulation occur following delivery, set in motion by the expansion of the lungs with the initial breaths of the newborn. The drop in pulmonary vascular resistance (PVR) that occurs with expansion of the lungs results in an increase in pulmonary blood flow and an elevation in the left atrial pressure. The increased oxygen tension in the blood normally results in involution of the ductus arteriosus. The elevated left atrial pressure
pushes septum primum against the edges of septum secundum, closing the foramen ovale. The result is separation of the pulmonary and systemic circulations. Abnormal cardiac development is frequently compatible with in utero existence because the fetal circulation allows one side of the heart to compensate for abnormal development of the contralateral side. Following delivery, significant malformations become evident. Most neonates, even those with congenital heart disease, have the ability to shunt blood across the foramen ovale, and ductal patency can be maintained with prostaglandin E1 (PGE1). Newborns with inadequate development of the right ventricular outflow tract, pulmonary valve, and main pulmonary artery, such as occurs with tetralogy of Fallot, can be managed by maintaining ductal patency. The patent ductus allows blood to shunt from the aorta to the pulmonary circuit and provides enough pulmonary blood flow to avoid lethal cyanosis. Similarly, the patient with severe coarctation and
arch hypoplasia is at risk for development of shock because of the obstruction of blood flow to the descending aorta. The use of PG-E1 maintains patency of the ductus arteriosus, allowing the right side of the heart to continue to provide flow to the descending thoracic aorta, preventing critical tissue ischemia, and relieving congestive heart failure from increased resistance to left-sided output. In this way, ductal patency can be manipulated in order to resuscitate neonates with a variety of cardiac lesions with either systemic obstruction or inadequate pulmonary blood flow. Emergent procedures are avoided, and patients are allowed to recover before intervention is undertaken. Figure 62-4 is a drawing of ductal-dependent pulmonary blood flow (Fig. 62-4a) and ductal-dependent systemic blood flow (Fig. 62-4b).
pushes septum primum against the edges of septum secundum, closing the foramen ovale. The result is separation of the pulmonary and systemic circulations. Abnormal cardiac development is frequently compatible with in utero existence because the fetal circulation allows one side of the heart to compensate for abnormal development of the contralateral side. Following delivery, significant malformations become evident. Most neonates, even those with congenital heart disease, have the ability to shunt blood across the foramen ovale, and ductal patency can be maintained with prostaglandin E1 (PGE1). Newborns with inadequate development of the right ventricular outflow tract, pulmonary valve, and main pulmonary artery, such as occurs with tetralogy of Fallot, can be managed by maintaining ductal patency. The patent ductus allows blood to shunt from the aorta to the pulmonary circuit and provides enough pulmonary blood flow to avoid lethal cyanosis. Similarly, the patient with severe coarctation and
arch hypoplasia is at risk for development of shock because of the obstruction of blood flow to the descending aorta. The use of PG-E1 maintains patency of the ductus arteriosus, allowing the right side of the heart to continue to provide flow to the descending thoracic aorta, preventing critical tissue ischemia, and relieving congestive heart failure from increased resistance to left-sided output. In this way, ductal patency can be manipulated in order to resuscitate neonates with a variety of cardiac lesions with either systemic obstruction or inadequate pulmonary blood flow. Emergent procedures are avoided, and patients are allowed to recover before intervention is undertaken. Figure 62-4 is a drawing of ductal-dependent pulmonary blood flow (Fig. 62-4a) and ductal-dependent systemic blood flow (Fig. 62-4b).
 FIGURE 62-1. Cardiac development from formation of the bulboventricular loop to the completely formed heart takes place from 30 to 49 days and is depicted here. (A) 30 days. The right and left ventricles are becoming evident on either side of the interventricular foramen, and septation of the ventricles has been initiated. At this point, the atrioventricular canal, the opening from the atria to the ventricular loop, communicates with the primitive left ventricle only. (B) 35 days. Differential growth results in a shifting of the atrioventricular canal so blood enters both the primitive right and left ventricle. The endocardial cushions are evident and will participate in septation of the ventricles, along with separation of the atrioventricular valve into the tricuspid and mitral valves. Septation of the outflow tract is initiated by swellings on the right and left sides of the conus and right superior and left inferior swellings on the truncus arteriosus. (C) 49 days. Separation of the ventricles and atrioventricular valves is completed, the conus and truncus have completed septation, the left ventricle ejects to the aorta, and the right ventricle ejects to the pulmonary artery. Congenital heart disease is nearly always the result of arrest of a portion of cardiac development. Defects of septation, such as atrial septal defects, ventricular septal defects, and atrioventricular septal defects, are caused by failure of any of the components of the developing atrial and ventricular septa; conotruncal truncal abnormalities result from failure of the outflow and proximal great vessels to complete septation and achieve proper alignment; and finally, lesions involving underdevelopment of the inflow or outflow of either ventricle will result in a spectrum of hypoplastic lesions of either the left or right heart. |
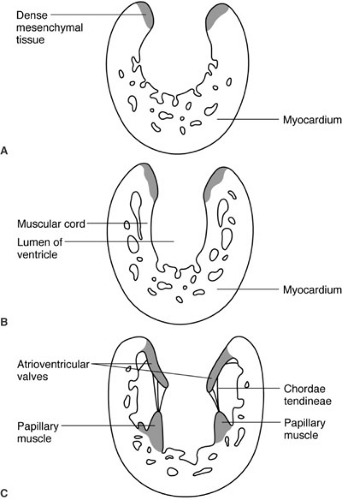 FIGURE 62-2. Formation of the valves is depicted. |
PRESENTATION AND DIAGNOSIS
Cardiovascular disease in the neonatal and pediatric age groups can present with congestive heart failure, cyanosis, or both and may progress to shock (defined as critically low levels of oxygen delivery to the tissues), resulting in anaerobic metabolism. Neonates with cyanotic congenital heart disease may have ductal-dependent pulmonary blood flow and may develop life-threatening hypoxemia when the normal spontaneous closure of the ductus arteriosus occurs. Neonates with left heart obstructive lesions depend on the patency of the ductus arteriosus for perfusion of the proximal and distal aorta. When spontaneous ductal closure occurs, this group of neonates will develop low cardiac output as a result of left heart obstruction. Therefore, the presenting symptoms in this group include poor perfusion, diminished peripheral pulses, decreased urine output, acidosis, and all signs and symptoms associated with shock.
The signs and symptoms of congestive heart failure during infancy include tachypnea, tachycardia, poor feeding, slow growth, cardiomegaly, and hepatomegaly. In infants with left-to-right shunt lesions, it is common for heart failure to present within the first few weeks of life rather than immediately after birth. In the presence of a shunting defect, PVR can remain elevated for the first several weeks of life. The eventual decrease in PVR results in increased left-to-right shunting with the corresponding development of congestive heart failure. Infants and children with congenital heart disease may be first identified by the presence of a characteristic heart murmur. Although some serious congenital heart defects presenting in the neonatal period or early infancy do not have specific murmurs, less life-threatening congenital cardiac defects such as pulmonary stenosis, aortic stenosis, and restrictive ventricular septal defects often present with loud murmurs in patients who are asymptomatic.
The physical examination continues to be the mainstay of diagnosis for congenital heart disease, and a systematic approach to the physical examination increases the likelihood that a correct diagnosis will be made and that important findings will not be missed. Initial observation for obvious characteristic facial features of a specific syndrome should be done with care. In addition, one should get a general impression of the color and level of comfort of the neonate or infant so a rapid assessment of stability can be made. Respiratory distress can be ruled out by simple observation. The precordium should be palpated. Cardiac hyperactivity, often present in neonates and infants with serious cardiac abnormalities, can be easily determined. Pulses should be palpated in all four extremities. Decreased pulses in all four extremities suggest low cardiac output and shock. Strong upper-extremity pulses, but weak or absent lower-extremity pulses, suggest coarctation of the aorta. Bounding pulses suggest aortic runoff into the pulmonary vascular bed, the result of either a patent ductus arteriosus or arteriovenous malformation. It is important to routinely measure blood pressure in all four extremities in order to quantify any differences.
 FIGURE 62-3. Fetal circulation. The circulation of the fetus following formation of the heart is depicted. The grayscale indicates the degree of desaturation of the blood. The darker shaded areas indicate more desaturated blood, whereas the white areas indicate more fully saturated blood. Umbilical venous return, the most saturated blood enters the inferior vena cava and is preferentially shunted across the patent foramen ovale to the left side of the heart to supply the cerebral circulation. Less fully saturated blood is pumped by the right ventricle through the ductus arteriosus to the descending thoracic aorta. Return to the placenta for gas exchange is via the umbilical arteries, which arise from the iliac arteries. |
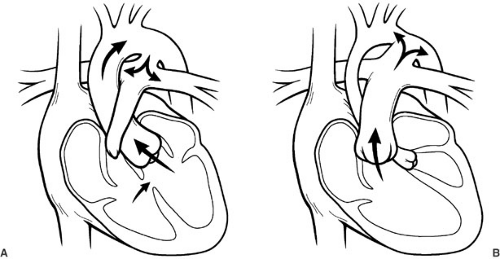 FIGURE 62-4. A patent ductus arteriosus can compensate for underdevelopment of either the right or left side of the heart. (A) Depicts ductal-dependent pulmonary blood flow in a case of pulmonary atresia and ventricular septal defect. Blood is shunted from the aorta to the pulmonary arteries to provide pulmonary blood flow. (B) Demonstrates ductal-dependent systemic flow in a patient with hypoplastic left heart syndrome. In this case, the single right ventricle ejects blood through the pulmonary root and systemic flow is supported by flow through the ductus arteriosus into the aorta. Flow to the head and coronary arteries is maintained via retrograde flow from the ductus. |
Abdominal palpation will allow one to determine the position of the liver and whether it is enlarged. In syndromes of left–right isomerism (heterotaxy), it is possible for the liver and stomach to be reversed within the abdomen. In addition, it is not unusual to encounter hepatomegaly in patients with heart failure. One should inspect the nail beds for cyanosis and for clubbing. Cyanosis can be detected visually only if at least 5 g of unsaturated hemoglobin per 100 mL of blood is present in the capillary bed. Thus, anemia may mask important cyanosis. Routine measurement of pulse oximetry is therefore important in all neonates, infants, and children who are evaluated for congenital heart disease.
Auscultation often yields information with regard to the anatomic diagnosis, as well as the specific hemodynamic aspects of a specific lesion. Auscultation should also be undertaken in a systematic fashion. One should routinely listen to the precordium over the apex of the heart, left lower sternal border, left upper sternal border, and right upper sternal border. At each area, one should note the first heart sound, systole, the second heart sound, and diastole. These areas typically represent events associated with the mitral valve, tricuspid valve, pulmonary valve, and aortic valve, respectively. In addition, locations between the previously mentioned sites should be auscultated, as should the back and midaxillary areas of the chest. For example, the presence of a ventricular septal defect is often manifested by a harsh, holosystolic murmur at the left lower sternal border.
Two-dimensional echocardiography with Doppler and color flow Doppler mapping has revolutionized the noninvasive diagnostic capabilities for determining spatial anatomy and function of the heart in pediatric cardiology (17,18). This modality, developed since the mid-1980s, has become an effective, accurate, and safe technique for the elucidation of anatomic and hemodynamic details of all congenital cardiac abnormalities. The two-dimensional study provides tomographic images of the heart. Images are obtained via directing of the ultrasound transducer beam along standard cross-sectional planes through the heart. Based on these standard views, an accurate diagnosis can be made of the most complex of congenital heart defects along with the detailed relationships of the intracardiac anatomy and function of the heart.
In addition, Doppler ultrasound has aided in the evaluation of the hemodynamic severity of a particular cardiac defect by evaluating the blood flow pattern within the heart and peripheral vessels. Doppler echocardiography will detect normal and abnormal blood flow patterns, and can quantify the severity of a blood flow disturbance. The principle of Doppler is based on the fact that the frequency of a transmitted sound is altered when the source of the sound is moving. Doppler echocardiography provides information about the velocity and direction of blood flow, making possible calculation of pressure gradients across areas of stenosis. The simplified Bernoulli equation: P1 – P2 = 4V22 (P1 – P2 = pressure gradient across the valve or defect in mm Hg; V2 = peak velocity across the valve or defect as measured by Doppler echocardiography in meters per second) can be used to estimate the pressure gradient across a stenotic valve, or a septal defect.
Diagnostic cardiac catheterization provides important anatomic and physiologic data. Vascular anatomy can be defined by cardiac catheterization and corresponding hemodynamic data obtained, such as pressure and flow data that are necessary to make determinations of vascular resistance. Indications for diagnostic cardiac catheterization include incomplete or inconclusive noninvasive anatomic diagnoses. Cardiac catheterization is uniquely suited for the determination of pulmonary artery architecture and pulmonary vascular resistance. The impact of pulmonary vasodilators on pulmonary vascular resistance is ideally determined in the catheterization laboratory. Before the development of echocardiography, cardiac catheterization was the mainstay of physiologic and anatomic assessment of congenital heart disease (19) and, although the role of cardiac catheterization has diminished as a purely diagnostic tool, the catheterization laboratory is now more and more the venue for intervention. Increasingly, the catheterization laboratory is the site for treatment of patients with patent ductus arteriosus, atrial and ventricular septal defects, and branch pulmonary artery stenosis.
Developing diagnostic modalities include cardiac ultrafast computed tomography (CT) and magnetic resonance imaging (MRI) currently available as an adjunct to echocardiography and cardiac catheterization. These newer imaging techniques provide excellent noninvasive images of the pulmonary arteries and pulmonary veins, anomalies of the systemic veins, and anomalies of the aortic arch. CT and MRI may be useful in the postoperative evaluation of patients and in cases where precise flow and pressure data are not necessary.
TREATMENT OF CONGENITAL HEART DISEASE
Treatment of congenital heart disease is primarily focused on (1) relief of congestive heart failure, defined as excessively high filling pressures, and frequently the result of excessive pulmonary blood flow due to defects in either the atrial or ventricular septum, or (2) the treatment of cyanosis due to either right-to-left shunt or failure of development of an in-series pulmonary and systemic circulation.
The modern era of care for patients with congenital heart disease began when Dr. Robert Gross successfully ligated the patent ductus arteriosus of a 7-year-old girl in August 1938 (20). This was followed by the successful repair of coarctation of the aorta by Clarence Crafoord in Sweden in 1944 and subsequently by Dr. Gross in the
United States (21). Gross had shown that surgery on the great vessels around the heart was possible. Helen Taussig, a pediatric cardiologist, made the observation that patients with tetralogy of Fallot and a patent ductus arteriosus had improved survival and functional status compared with patients with tetralogy of Fallot and no patent ductus. She conjectured that for patients with cyanotic heart disease the addition of a ductus arteriosus would result in relief of cyanosis. Dr. Taussig found a receptive ear to her theories in Alfred Blalock. In 1944, Dr. Blalock performed the first systemic to pulmonary artery shunt in a patient with tetralogy of Fallot (22). This procedure was based on the largely unheralded work of his laboratory assistant Vivien Thomas. The Blalock-Taussig shunt was the first example of a novel anatomic solution to a physiologic problem. In 1952, Muller and Dammann reported pulmonary artery banding for the palliation of patients with congestive heart failure due to excessive pulmonary blood flow (23). In the 14 years between 1938 and 1952, fundamental steps had been taken in the therapy of congenital heart disease. Pathologic anatomy had been both corrected (ligation of a patent ductus arteriosus) and palliated. Patients with inadequate pulmonary blood flow could be managed by creation of a Blalock-Taussig shunt and patients with congestive heart failure (acyanotic congenital heart disease) could be managed with a pulmonary artery band. The development of these palliative procedures lead to the first classification of congenital heart disease into cyanotic and acyanotic lesions. This classification directed the initial palliative procedure. Blue babies were shunted; babies with heart failure underwent pulmonary artery banding.
United States (21). Gross had shown that surgery on the great vessels around the heart was possible. Helen Taussig, a pediatric cardiologist, made the observation that patients with tetralogy of Fallot and a patent ductus arteriosus had improved survival and functional status compared with patients with tetralogy of Fallot and no patent ductus. She conjectured that for patients with cyanotic heart disease the addition of a ductus arteriosus would result in relief of cyanosis. Dr. Taussig found a receptive ear to her theories in Alfred Blalock. In 1944, Dr. Blalock performed the first systemic to pulmonary artery shunt in a patient with tetralogy of Fallot (22). This procedure was based on the largely unheralded work of his laboratory assistant Vivien Thomas. The Blalock-Taussig shunt was the first example of a novel anatomic solution to a physiologic problem. In 1952, Muller and Dammann reported pulmonary artery banding for the palliation of patients with congestive heart failure due to excessive pulmonary blood flow (23). In the 14 years between 1938 and 1952, fundamental steps had been taken in the therapy of congenital heart disease. Pathologic anatomy had been both corrected (ligation of a patent ductus arteriosus) and palliated. Patients with inadequate pulmonary blood flow could be managed by creation of a Blalock-Taussig shunt and patients with congestive heart failure (acyanotic congenital heart disease) could be managed with a pulmonary artery band. The development of these palliative procedures lead to the first classification of congenital heart disease into cyanotic and acyanotic lesions. This classification directed the initial palliative procedure. Blue babies were shunted; babies with heart failure underwent pulmonary artery banding.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


