Congenital Disorders of Glycosylation
Jaak Jaeken
Glycosylation is an important posttranslational protein modification occurring in the cytoplasm, the endoplasmic reticulum, and the Golgi apparatus. A rapidly growing family of genetic diseases is due to defects in protein glycosylation (congenital disorders of glycosylation [CDG]). Most CDG are severe, multisystem diseases with important neurological involvement. Some 30 CDG have been identified. CDG due to an N-glycosylation defect (there are 18 disorders) comprise two groups: CDG-I (with absence of one or more glycans; CDG-Ia through CDG-IL) and CDG-II (with incomplete glycans; CDG-IIa through CDG-IIf). Six disorders have been identified in O-glycosylation, including some long-known diseases such as hereditary multiple exostoses; another six disorders have a combined N- and O-glycosylation defect. Important tools in the diagnosis are transferrin isoelectric focusing, analysis of lipid-linked oligosaccharides and of protein-linked glycans, and mutation analysis.
PROTEIN GLYCOSYLATION
Congenital disorders of glycosylation (CDG) are a rapidly growing family of genetic diseases caused by defects in the synthesis of the glycan moiety of glycoconjugates (glycoproteins and gly-colipids). There are two main types of protein glycosylation: N-glycosylation and O-glycosylation. N-glycosylation (N-glycans attached to an amino group of asparagine of proteins) comprises an assembly part and a processing part and extends over three cellular compartments: the cytosol, the endoplasmic reticulum (ER), and the Golgi.
The assembly part of the N-glycosylation starts on the cytosolic side of the ER, with the transfer of N-acetylglucosamine (GlcNAc) phosphate from UDP-GlcNAc to membrane bound dolichyl monophosphate (Dol-P), forming GlcNAc-pyrophosphate-dolichol (GlcNAc-PP-Dol). One GlcNAc and five mannose (Man) residues are subsequently attached to this lipid-linked monosaccharide in a stepwise manner (Fig. 163-1). The donor of these mannoses is a nucleotide-activated sugar, GDP-Man, which is synthesized from fructose 6-phosphate, an intermediate of the glycolytic pathway (Fig. 163-2). The lipid-linked hep-tasaccharide Man5 GlcNAc2 is translocated by a flippase across the ER membrane and is elongated at the lumenal side by the attachment of four mannose residues and subsequently of three glucose residues. The four mannosyl-transferases and three glucosyltransferases involved require dolichyl-phosphate-bound monosaccharides (Dol-P-Man and Dol-P-Glc). The completed Glc3 Man9 GlcNAc2 oligosaccharide is then transferred to selected asparagine residues of the nascent proteins by the oligosaccharyltransferase complex.
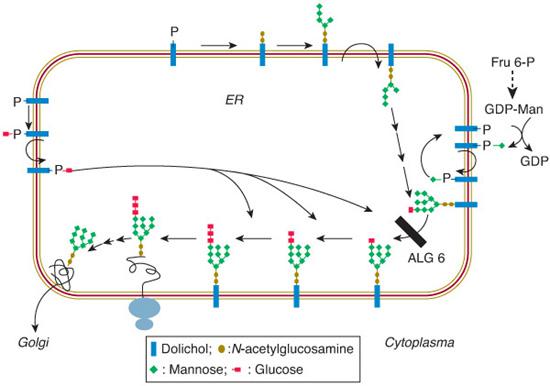
FIGURE 163-1. Scheme of the endoplasmic reticulum part of the N-glycosylation pathway (see text for explanation). The black bar beside ALG6 indicates the defect in CDG-Ic (ALG6 or glucosyltransferase I defect).
The processing part of the N-glycosylation starts in the ER by trimming the glucoses (catalyzed by glucosidases I and II) and one mannose (catalyzed by α-mannosidase I). The residual glycoprotein intermediate is directed to the cis-Golgi, where the processing pathway branches. A minor branch targets glycoproteins to the lysosomes (after the action of a GlcNAc-phosphotransferase and removal of the GlcNAc residues, leaving high-mannose glycoproteins capped with Man 6-P). The main branch leads to further trimming man-noses (leaving a trimannosyl core) and the addition of GlcNAc, galactose, and eventually, sialic acid, in the medial- and trans-Golgi. Another modification of many N-glycoproteins in the Golgi is the attachment of fucose to the GlcNAc residue that is linked to asparagine.
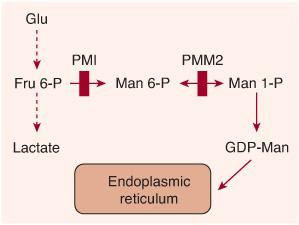
FIGURE 163-2. Scheme of the synthesis of guan-osine diphosphate (GDP)-mannose from fructose 6-phosphate. Vertical red bars indicate defects in CDG-Ia (PMM2 defect) and in CDG-Ib (PMI defect).
O-glycosylation (O-glycans attached to the hydroxyl group of threonine or serine of proteins) has no processing part and thus consists only of assembly. Unlike N-glycosylation, this assembly mainly occurs in the Golgi. O-glycan structures show a greater diversity than N-gly-cans. Examples of important O-glycans are O-N-acetylgalactosaminylglycans (mucin-type glycans), O-xylosylglycans (glycosaminylglycans), O-mannosyl glycans, and O-fucosylglycans.
GENETIC DISEASES OF PROTEIN N-GLYCOSYLATION
Eighteen diseases are known in protein N-glycosylation: 14 assembly defects (CDG-I group), designated CDG-Ia through CDG-In, and four processing defects (CDG-II group), designated CDG-IIa through CDG-IId. We discuss only CDG-Ia, CDG-Ib, and CDG-Ic in some detail, since all the other diseases are very rare. 
PHOSPHOMANNOMUTASE2 DEFICIENCY (CDG-IA)
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
CDG-Ia is by far the most frequent protein N-glycosylation disorder (some 600 patients known). The clinical spectrum is very broad: The nervous system is affected in all patients, and most other organs are involved in a variable way. The neurological picture comprises alternating internal strabism and other abnormal eye movements, axial hypotonia, psychomotor retardation, ataxia, and hyporeflexia. After infancy, symptoms include retinitis pigmentosa, stroke-like episodes, and sometimes epilepsy. During the first year(s) of life, there are variable feeding problems (anorexia, vomiting, diarrhea) that can result in severe failure to thrive. Other features are a variable dysmorphy (large, hypoplastic/dysplastic ears, abnormal subcutaneous adipose-tissue distribution [fat pads, inverted nipples]), mild to moderate hepatomegaly, skeletal abnormalities (including atlantoaxial subluxation), and hypogonadism. Some infants develop pericardial effusion or cardiomyopathy. At the other end of the clinical spectrum are patients with a very mild phenotype (no dysmorphy, slight psychomotor retardation). Patients often have an extroverted and happy appearance.
 METABOLIC DERANGEMENT
METABOLIC DERANGEMENT
Phosphomannomutase2 (PMM2) deficiency is a (cytosolic) defect in the second step of the mannose pathway (transforming mannose-6-phosphate into mannose-1-phosphate), which normally leads to the synthesis of guanosine diphosphate (GDP)-mannose (Fig. 163-2). This nucleotide sugar is the donor of mannose used in the ER to assemble the dolichol-pyrophosphate oligosaccharide precursor. Deficiency of GDP-mannose causes hypoglycosylation of numerous glycoproteins, including serum proteins, lysosomal enzymes, and membranous glycoproteins.
 GENETICS
GENETICS
CDG-Ia (OMIM 212065) is an autosomal-recessive disease due to mutations of PMM2 on chromosome 16p13. At least 80 mutations have been identified (mainly missense mutations), the most frequent being the R141H mutation. Prenatal diagnosis is possible by enzymatic analysis of amniocytes and chorionic villus cells; this should be combined with mutation analysis of the PMM2 gene.
 DIAGNOSTIC TESTS
DIAGNOSTIC TESTS
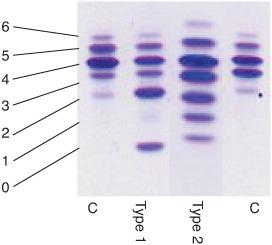
The diagnosis of CDG-Ia (and of congenital disorders of N-glycosylation in general) is usually made by isoelectrofocusing (IEF) and immunofixation of serum transferrin (Fig. 163-1). Normal serum transferrin is mainly composed of tetrasialotransferrin and small amounts of mono-, di-, tri-, penta-, and hexasialotransferrins. The partial deficiency of sialic acid (a negatively charged and end-standing sugar) in CDG causes a cathodal shift. Two main types of cathodal shift can be recognized: Type 1 is characterized by an increase of both disialo- and asialotransferrin and a decrease of tetrasialotransferrin; in type 2 there is also an increase of the tri- and/or monosial-otransferrin bands (Fig. 163-3). In PMM2 deficiency, a type 1 pattern is found. Very recently, capillary zone electrophoresis of total serum has been introduced for the diagnosis of CDG (Fig. 163-1). In addition to the previously mentioned serum glycoprotein abnormalities, laboratory findings include elevation of serum transaminase levels, hypoalbuminemia, hypocholesterolemia, and tubular proteinuria. To confirm the diagnosis, the activity of PMM2 should be measured in leukocytes or fibroblasts.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


