Congenital Diaphragmatic Hernia
INTRODUCTION
Congenital diaphragmatic hernia (CDH) is a developmental defect in the diaphragm associated with herniation of the abdominal viscera and ipsilateral pulmonary hypoplasia most pronounced on the ipsilateral side. In the current era, approximately 64% of cases in the United States will be diagnosed prenatally by midtrimester ultrasound, and the majority of the remaining newborns will present in the first few hours of life with varying degrees of respiratory distress. Herniation occurring later in gestation is associated with less-significant pulmonary hypoplasia and milder respiratory distress in the newborn period and is often not prenatally diagnosed. Prenatal diagnosis and advances in neonatal care have improved survival to 75% for isolated CDH. However, newborns with more severe CDH and those with other congenital anomalies or chromosomal defects continue to have a significant risk of death and morbidities, such as chronic lung disease, pulmonary hypertension, gastroesophageal reflux, failure to thrive, hernia recurrence, chest wall deformities, scoliosis, and adverse neuro-developmental outcome.
A voluntary international registry for CDH infants was established in 1995 by Dr. Kevin Lally to collect data on infants with CDH with the goal of advancing the care and outcome for such infants.1 The CDH Study Group Registry now includes data from over 6000 patients from 93 centers in 10 countries and has provided important information regarding the efficacy of various therapies and predictors of outcome.
EPIDEMIOLOGY
The birth prevalence of CDH in population-based studies ranges from 0.17 to 0.57 per 1000 live births.2,3 CDH is more frequent in males, with a ratio of 1.4 to 1, and less frequent among African Americans.3 Associated congenital anomalies occur in 25%, abnormal karyotype in 16%, and genetic syndromes in 5%. Cardiac malformations are the most frequent congenital anomalies seen in the CDH population, followed by anomalies of the ribs and sternum, brain, and spine. Chromosomal abnormalities seen include trisomy 18, 13, 21, 22, and 9 as well as other nontrisomy chromosomal anomalies.3 Fryns syndrome is the most common autosomal recessive syndrome seen in the CDH population, occurring in 1% to 10%, and is associated with a high mortality rate and extremely poor quality of life.4
PATHOPHYSIOLOGY
The diaphragm is formed by fusion of the septum transversum and the pleuroperitoneal folds around the eighth week of gestation, thereby separating the pleural cavities from the peritoneal cavity. Failure of this closure will result in a diaphragmatic hernia, which most frequently appears in the posterior/lateral part, referred to as a Bochdalek hernia. Hernias in the anterior part of the diaphragm, known as Morgagni hernias, are less frequent. About 85% to 90% of diaphragmatic hernias occur on the left side and 10% to 15% on the right side. Bilateral hernias are uncommon (1%-2%). Most commonly, the defect is complete with a direct communication between the pleural and peritoneal cavities. In about 10% to 15%, however, the defect is covered by a thin membrane, or hernia sac, consisting of the pleural and peritoneal membranes.5,6
Failure of diaphragmatic closure occurs in the transition between the embryonic phase and the pseudoglandular phase of lung development. Subsequent lung development is characterized by a reduction in the number of airway generations, resulting in a decreased number of terminal bronchioles and alveoli. Alveolar septa are thickened, and the general histologic appearance is that of an immature lung. The number of arterial generations is also decreased, and the pulmonary arteries have a thicker muscle layer than normal. Furthermore, muscularized arterioles are seen more peripheral than in the normal lung. The weight, DNA, and protein content of the lungs are decreased. The pulmonary hypoplasia affects both lungs but is more severe on the ipsilateral side.5,6
During fetal life, the lungs are a liquid-producing organ and are the main producers of amniotic fluid. This occurs through an active transport of chloride ions across the alveoli. The ion transporter NKCC-1 (Na+-K+-2Cl– cotransporter) is localized in the basolateral membranes of the alveolar cells and is considered rate limiting for this process. Fetal lung liquid production is essential for lung growth.7 Around birth, the lungs instead change to become a water-reabsorbing organ, which occurs through active sodium transport through the ion channel ENaC (epithelial sodium channel) in the alveolar cells. In recent experiments, the expression of ENaC in rat lungs with experimentally induced CDH was reduced,7,8 and in newborns with CDH, ENaC was reduced in tracheal aspirates compared to controls.9 This fits well with the clinical observation that newborn babies with CDH need more frequent airway suctioning than other newborns and indicates that, in addition to hypoplasticity, CDH lungs are immature.
The etiology of the diaphragmatic defect and the concomitant pulmonary hypoplasia seen with CDH is unknown. Classically, the primary defect was considered to be the failure of diaphragmatic closure, allowing the abdominal viscera to herniate into the thoracic cavity, thereby interfering with pulmonary development and causing pulmonary hypoplasia. Recent data, however, have supported a more complex mechanism mainly based on concomitant abnormalities in diaphragmatic and pulmonary development.10–12 An interesting combination is the “dual-hit hypothesis”13 explaining the pathogenesis of pulmonary development by 2 different insults. The first event, caused by unidentified genetic and environmental factors, occurs early in development before normal diaphragmatic closure, and the second insult is caused by interference from the herniated abdominal viscera.
In the fetus, the pulmonary vascular resistance is normally high, and blood is shunted from the pulmonary artery through the ductus arteriosus to the aorta. In healthy babies, the vascular resistance drops after birth, leading to a rapid fall in pulmonary artery pressure, change of direction in ductal shunt, and eventually closure of the ductus. Probably due to decreased cross-sectional area and increased musculature of the pulmonary vascular bed, the pulmonary vascular resistance remains high in patients with CDH and pulmonary hypoplasia, which in severe cases will lead to suprasystemic pressure in the pulmonary artery and to right-to-left shunting at the level of the ductus and foramen ovale. Decreased blood flow through the lungs results in hypoxemia, hypercarbia, and acidosis and initiates a vicious cycle, as hypoxia and acidosis are potent stimuli for the pulmonary artery smooth muscle cells to contract, which further increases pulmonary vascular resistance. Other important stimuli that cause increased pulmonary vascular resistance are hypothermia and stress.5,14–16 Severe and progressive respiratory failure will ensue unless the vicious cycle is broken.
The majority of newborns with CDH will present within 24 hours after birth with respiratory distress. The more severe the pulmonary hypoplasia is, the earlier the symptoms will present. The most severe cases will need immediate ventilator support and intensive care directly after birth. By contrast, mild cases can occasionally present later with diffuse respiratory symptoms or be diagnosed by incidental chest radiograph. In rare instances, the bowel can be herniated through a small diaphragmatic defect later in life, leading to an incarcerated hernia with classical signs of intestinal obstruction.
DIFFERENTIAL DIAGNOSIS
Most CDH cases are diagnosed prenatally by ultrasound or postnatally by chest radiograph (Figure 27-1). The differential diagnosis includes diaphragmatic eventration, congenital cystic adenomatoid malformation (CCAM), bronchopulmonary sequestration, bronchogenic cysts, bronchial atresia, enteric cysts, and teratomas. Definitive prenatal sonographic diagnosis is made when abdominal organs are present in the fetal chest. Right-sided CDH is more frequently missed or misdiagnosed, as liver can have a similar echogenicity to lung.
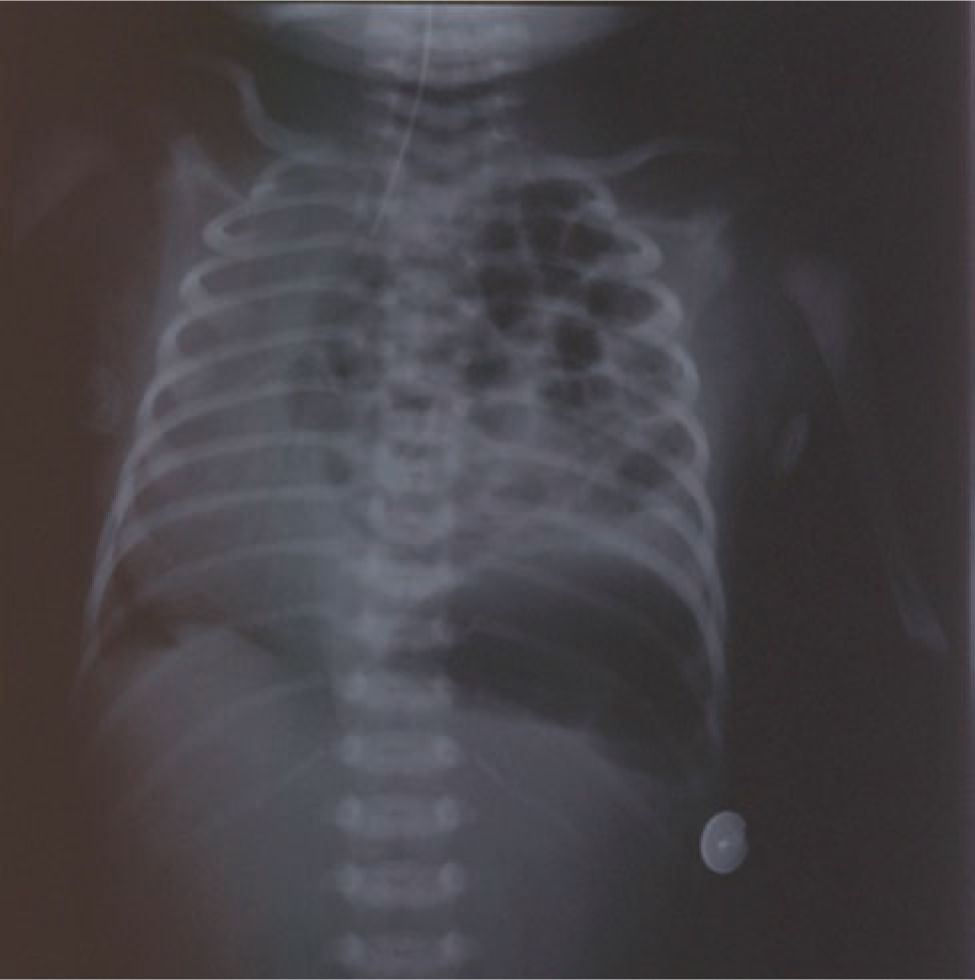
FIGURE 27-1 Postnatal chest radiograph demonstrating a newborn with a left congenital diaphragmatic hernia (CDH) with dilated loops of bowel in the left hemithorax and shift of the heart to the right.
DIAGNOSTIC TESTS
Prenatal ultrasonography (US) will identify most fetuses with CDH. Diagnosis has been described as early as the 11th week of gestation, and in many developed countries, CDH is diagnosed during routine ultrasound screening during the first part of the second trimester. Mediastinal shift and abdominal organs within the thoracic cavity will be the hallmark. In left-sided hernias, the mediastinum is shifted to the right, and intestines, sometimes together with the stomach, and the left liver lobe are found in the thorax. In right-sided hernias, often the liver is herniated. In a strict 4-chamber view, the cross-sectional area of the contralateral lung can be calculated, and when this is divided by the head circumference measured at the standard biparietal view, the lung-to-head ratio (LHR) is obtained. In left-sided hernias, this ratio is felt to correlate with outcome. As the LHR normally varies during gestation, a better prognostic parameter is the observed LHR divided by the expected LHR at that time point of gestation. US can also identify the position of the liver in left-sided hernias, which influences prognosis.17,18
Prenatal magnetic resonance imaging (MRI) assessment is used more frequently in the current era. Besides confirming the diagnosis and position of the liver, it allows for a more accurate estimation of lung volume with subsequent correlation to outcome. In cases with equivocal US findings, MRI will also be of value.
Postnatally, classical signs at physical examination of a newborn with CDH are respiratory distress in conjunction with a scaphoid abdomen. In left-sided hernias, the heart sounds will be heard more clearly to the right. Breath sounds, on the other hand, are easily transmitted over the chest and can be fairly symmetrical despite unilateral lung hypoplasia.
The diagnosis of CDH is confirmed by a plain chest radiograph, which demonstrates a mediastinal shift to the contralateral side and bowel loops in the thorax. Air is frequently seen in the esophagus. Rarely, the radiograph can mimic that of a patient with CCAM involving the left lower lobe. However, with CCAM, both diaphragms can be visualized on the lateral view. In doubtful cases, contrast medium given in the gastric tube can be useful. Computed tomographic (CT) scan is rarely needed for diagnosis.
MANAGEMENT
Prenatal Evaluation, Counseling, and Planning for Delivery
Once CDH has been diagnosed in utero, it is important to determine if the defect is isolated or associated with other congenital or chromosomal anomalies known to affect outcome. Level 2 US to screen for other anatomic abnormalities should be performed, and an amniocentesis should be considered to identify chromosomal abnormalities. After this evaluation, the medical team is faced with presenting the various options to the parents. At most centers, the options include pregnancy termination or delivery at a tertiary-level center with multimodality support available. Pregnancy termination may or may not be an option depending on the gestational age at the time of diagnosis. Fetal surgery is now offered to selected prenatally diagnosed infants at a few centers.
Fetal Therapy
In a classical work performed by Harrison et al, a balloon was inserted into 1 pleural cavity in fetal lambs during the third trimester. If the balloon was left inflated, the lambs died soon after birth and had pulmonary hypoplasia much resembling what is seen in human CDH. In another group of sheep, the balloon was instead deflated after 20 days. The lungs were better developed in these lambs compared to the previous group.19 These preliminary experiments were followed by others in which a diaphragmatic defect was surgically created in fetal lambs during the first trimester. Typical signs of CDH with pulmonary hypoplasia were seen after birth. If the defect was surgically repaired during the second trimester, the pulmonary hypoplasia was significantly less pronounced. Success in large-animal studies led to attempts in humans with prenatally diagnosed CDH.
However, the results from human open repair of CDH in utero were disappointing. Of 14 patients with isolated left-sided CDH, there were only 4 survivors. The major problem was technical, as it was not possible to reduce the herniated liver into the abdomen without kinking the umbilical vein. The investigators concluded that open fetal repair was not possible in the group of left-sided hernia patients with the worst prognosis (ie, those with herniated liver). Following this, a prospective study was conducted in which patients with nonherniated livers were randomized to open fetal repair or conventional therapy. The study failed to demonstrate any difference in outcome. Open fetal repair has since been abandoned.20
In contrast to patients with CDH, the lungs of fetuses with rare malformations leading to congenital high-airway obstruction syndrome are hyperplastic. During fetal life, the lungs produce fluid through active excretion of chloride ions, and the lung water escapes through the trachea to the amniotic fluid when the glottis periodically opens. High-airway obstruction prevents escape of fluid and causes an increased airway pressure in the fetus, leading to stimulated lung growth. This has been extensively studied in animal experiments. The hyperplastic lungs exhibit a normal histologic architecture and consist of a larger number of alveoli and capillaries. Experimentally performed tracheal obstruction (TO) in the fetal lamb model enhances lung growth, abdominal viscera are reduced to the abdominal cavity, and postnatal lung function is improved. However, type II pneumocytes producing surfactant are decreased after TO, but this effect may be reduced if TO is discontinued prior to birth.
Tracheal obstruction in human fetuses with CDH was first performed by Harrison et al in 199621 by maternal laparotomy, hysterotomy, and open fetal neck dissection. A special technique called the ex utero intrapartum treatment (EXIT) procedure was developed for delivery. The mother was anesthetized and the uterus relaxed. The fetal head and neck were mobilized through a hysterotomy, and the fetal airway was secured before the placental circulation was interrupted. Despite stimulated lung growth, the mortality after TO was initially high, which mainly was attributed to complications from preterm labor.
Improved techniques and the development of smaller laparoscopic instruments enabled the fetal endoscopic (FETENDO) surgical procedure, in which laparoscopic trocars are placed in the uterus under ultrasound guidance. Subsequently, intratracheal balloons were developed, making the technique less invasive and decreasing the risk for tracheal and recurrent laryngeal nerve damage. The results were encouraging in comparison to historical controls, but in a randomized controlled trial, the survival of fetuses with left-sided CDH, liver-up, and LHR less than 1.4 treated with FETENDO was 73%, which was equivalent to the control group.22
A technique for complete percutaneous TO has subsequently been developed by Deprest and coworkers23 using a small fetoscope and intratracheal balloons. With this method, which currently is practiced in some European centers, it is possible to remove the TO before birth so that the babies can be delivered normally. Again, significantly higher survival rates have been published compared to historical controls, but results from randomized controlled trials have not yet been published.
Postnatal Medical Management
The antenatal diagnosis of CDH allows the immediate neonatal care of infants with CDH to be optimized. Not only are parents able to be educated regarding the diagnosis and treatment, but also the delivery of medical and surgical care can be coordinated between perinatology, neonatology, and pediatric surgical services. Birth at a tertiary center with pediatric surgery, neonatology, and the availability of advanced therapies such as high-frequency ventilation, inhaled nitric oxide, and extracorporeal membrane oxygenation (ECMO) is desirable. Two studies performed by the Canadian Neonatal Network found a survival benefit for newborns with CDH who were delivered at high-volume tertiary centers.24,25
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


