Congenital Diaphragmatic Hernia
Kevin P. Lally
Division of Pediatric Surgery, University of Texas Health Sciences Center, Houston, Texas 77030.
INTRODUCTION
Congenital diaphragmatic hernia (CDH) is a rare congenital anomaly characterized by a defect in diaphragm development with subsequent herniation of abdominal contents into the thorax. The two most prevalent types are herniation anteriorly in the diaphragm (Morgagni hernia) and posterolateral (Bochdalek-type) hernias. The incidence of posterolateral CDH has been reported from between 1 in 2,000 and 1 in 5,000 live births (1). The true incidence is higher because some fetuses with severe multiple anomalies, including CDH, do not survive to birth because the pregnancy is spontaneously aborted. Many of the fetuses who survive to delivery have significant lung hypoplasia and other anomalies. The lung hypoplasia and associated anomalies lead to a high mortality and long-term morbidity. In addition to the high inherent mortality (CDH accounts for more than 1% of total infant mortality in the United States), CDH ranks among the more costly of correctable conditions, with an estimated cost per new case of $250,000, and an overall estimated yearly cost of $264,000,000 in the United States (1995 dollars) (2).
The relative rarity of CDH prevents the conduct of well-designed clinical studies at single centers. As a result, the management of CDH has evolved through several eras, each based largely on retrospective reviews from centers with varying numbers of patients. In the past few years, a number of centers have collaborated to collect multiinstitutional data on CDH. More recently, these multiinstitutional data from more than 1,000 neonates have been used to categorize patients into low- and high-risk groups (3). This may allow for better comparisons between centers and potentially for stratification of patients for future studies.
History
Riverius recorded the first congenital diaphragmatic hernia in 1679 (4). A posterolateral defect was shown at autopsy of an infant who died in 1701 by Holt. Morgagni described several types of diaphragmatic hernia in 1761, including the anterior hernia defect now associated eponymously with him. There were scattered reports of CDH in the nineteenth century, including one by Victor Bochdalek, who described two infants with posterolateral defects found at autopsy. This posterolateral diaphragmatic defect is now known as a Bochdalek hernia. Although there were some attempts at repair of this defect throughout the late nineteenth and early twentieth century, the first successful repair of a posterolateral CDH in a child was reported by Heidenhain in 1905. Hedblom first suggested that early intervention and reduction of the hernia might improve survival and proposed the paradigm that visceral herniation and lung compression led to pulmonary hypoplasia (5). In 1940, Ladd and Gross reported 9 of 16 survivors, and in 1946, Gross reported the first survivor repaired at less than 24 hours of age (6). Operative mortality for neonates was high during this time, and many surgeons recommended delaying repair until the child was older; however, regardless of management, most of these infants succumbed from respiratory failure. Gross recommended that repair should not be delayed and that it should occur immediately. This policy led to attempts at earlier repair with the intent of relieving a tension effect of the intestines in the thorax. By the 1980s, CDH was considered one of the most urgent of surgical emergencies. Survival at that time was quoted at around 50%, despite advances in neonatal critical care. These data were misleading, however, because many patients were now surviving transfer to tertiary facilities, whereas they would have died in the past. During the 1980s and 1990s, there were many advances in neonatal respiratory care, including the availability of extracorporeal membrane oxygenation (ECMO), high-frequency oscillation, surfactant, and inhaled nitric
oxide (NO). Although all these therapies have been used in infants with CDH, as discussed here, evidence of effectiveness has been lacking for many.
oxide (NO). Although all these therapies have been used in infants with CDH, as discussed here, evidence of effectiveness has been lacking for many.
Diaphragm and Lung Development
The posterolateral defect in the diaphragm was originally believed to represent a failure of the pleuroperitoneal canal to close, but more recent information supports a different concept. Greer and others showed that the problem may arise within the pleuroperitoneal fold which gives rise to the diaphragm (7). Using an animal model, Babiuk et al. found no evidence that the musculature for the diaphragm originates from the septum transversum or the lateral body wall. Rather, cellular muscle precursors migrate from cervical somites and populate the diaphragm (8). The mesenchymal substrate that forms the diaphragm likely originates from the somatopleure. Knockout mice for c-met receptors (which bind HGF/SF, a chemoattractant for guiding muscle precursor cells) show formation of an amuscular diaphragm.
Much of critical organogenesis in the fetus occurs during the same time period as diaphragm development. Cardiac and lung formation occur during the third and fourth weeks of gestation. Lung development is discussed elsewhere (see Chapter 11), but the major components of the conducting airways have developed by the sixteenth week of gestation. Development of respiratory bronchioles and alveoli continues until well after birth. Lung development in infants with CDH is impaired early enough in gestation that there is a smaller number of bronchial divisions and alveoli. Some researchers have suggested that the primary problem in patients with CDH is pulmonary hypoplasia with secondary diaphragm maldevelopment (9). The etiology of the lung hypoplasia is unclear at this time; however, a number of the genes responsible for lung development have altered expression in a murine model of CDH (10). Alveolar growth and pulmonary vasculature are linked. The importance of this observation is that the pulmonary vasculature in infants with CDH is also abnormal. There is clear evidence of a smaller vascular tree as well as abnormal muscularization of the arterioles in the gas-exchanging portions (respiratory bronchioles and distal airways) of the lung. This likely contributes to the significant pulmonary hypertension seen clinically in these patients.
During the eighth to tenth week of gestation, the developing extracoelonic intestine normally returns to the abdomen. With return of the bowel, a defect in the diaphragm allows the intestine to enter the thorax. The pulmonary hypoplasia demonstrable in animal models of CDH can be modulated, depending on the timing and amount of intestine herniated into the thorax. This observation may be relevant in infants with CDH because patients with a large anatomic defect have a significantly higher morbidity and mortality rate compared with infants with a small defect (11).
As mentioned previously, critical organogenesis occurs during the same general time frame as CDH development. This includes cardiac development and is consistent with the clinical observation that 20% to 25% of infants with CDH have associated congenital heart disease. It is unclear whether there is a specific field defect in the mesoderm in some of these patients or whether the defects occur as a result of a teratogen (10). The defects may range from a smaller than average left ventricle to a variety of major anomalies, including hypoplastic left heart. Presence of significant cardiac anomalies, is associated with a much worse survival in patients with CDH (12).
Posterolateral Hernia
Diagnosis
Classically, infants with CDH present in respiratory distress either at birth or within the first few hours of life. Currently, between 40% and 60% of infants with CDH are diagnosed prenatally (13).
The diagnosis of congenital diaphragmatic hernia is suspected when the prenatal ultrasound demonstrates the heart in an abnormal location and fluid-filled loops of bowel are visualized in the thorax (Fig. 58-1). The diagnosis can be confused with a cystic adenomatoid malformation of the lung, and a fetal magnetic resonance imaging may be helpful to distinguish the two (14) (Fig. 58-2).
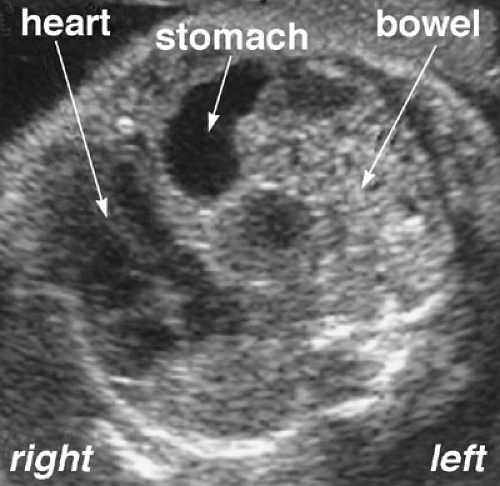 FIGURE 58-1. Prenatal ultrasound of a fetus with diaphragmatic hernia. At the level of the four-chamber heart view, the stomach and bowel can be clearly visualized. |
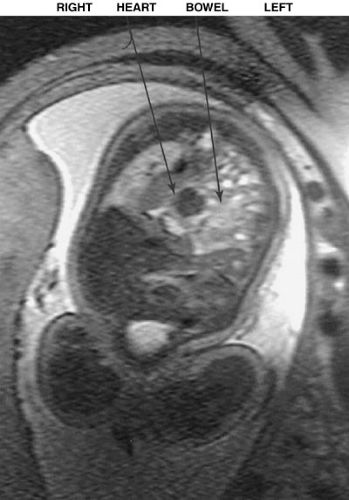 FIGURE 58-2. Prenatal MRI of a fetus with a left-sided diaphragmatic hernia. The heart can be seen pushed to the right, and the bowel is in the left thorax. |
In infants who present at or after delivery, respiratory distress may be due to underlying pulmonary hypoplasia or from compression, over hours or occasionally days, of the lung from gas-filled loops of bowel. The infant will often have a scaphoid abdomen (Fig. 58-3). A chest radiograph will usually show gas-filled loops of bowel in the chest with a shift of the heart away from the side of the defect side (Fig. 58-4). Eighty percent of infants with CDH have a left-sided defect, and 19% right, but whether left or right, the heart is shifted to the contralateral side. One percent of patients have bilateral herniae. The prognosis for these patients is much worse than for those with unilateral hernia (15). There may be bowel sounds in the chest, and the cardiac exam will show the shift of the heart to the side opposite the hernia. As for prenatal diagnosis, there can occasionally be confusion with a cystic adenomatoid malformation of the lung in newborns. Location of the nasogastric tube in the thorax on radiograph or an upper gastrointestinal contrast study will aid in confirming the diagnosis.
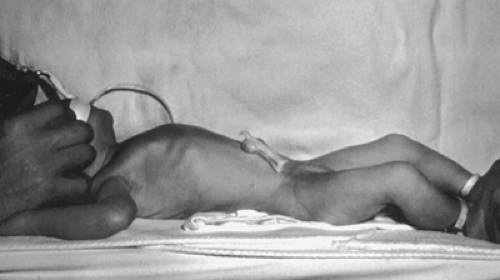 FIGURE 58-3. A newborn with a congenital diaphragmatic hernia. Note the scaphoid abdomen. |
The majority of infants with CDH will develop symptoms within the first 24 hours of life, although symptoms can be manifest at older ages and sometimes even at several months of age or later. Infants presenting with symptoms later may have feeding difficulty, respiratory problems, or bowel obstruction from an incarcerated hernia. In older patients, an upper gastrointestinal contrast study is often of value in establishing the diagnosis (16).
MANAGEMENT
Prenatal Management
Because a large number of infants with CDH are now diagnosed in utero, the option to treat the patient before delivery is available. CDH has a significant mortality rate and long-term morbidity, and this mortality rate has spurred the hope that in utero intervention may improve outcome. One of the difficulties in evaluating the risks and benefits of prenatal intervention is the inability to determine
which fetus is at the highest risk of death. Use of the ratio of lung to head or lung to thorax size, and even blood flow velocity as determined by ultrasound have been suggested (17,18). None of these has been validated in a large series at present, and many are observer dependent. At present, the problem of correctly stratifying a heterogenous group of patients and offering prenatal therapy to the selected high-risk fetus has not been resolved. Suggested methods to treat the fetus have included both medical and surgical approaches.
which fetus is at the highest risk of death. Use of the ratio of lung to head or lung to thorax size, and even blood flow velocity as determined by ultrasound have been suggested (17,18). None of these has been validated in a large series at present, and many are observer dependent. At present, the problem of correctly stratifying a heterogenous group of patients and offering prenatal therapy to the selected high-risk fetus has not been resolved. Suggested methods to treat the fetus have included both medical and surgical approaches.
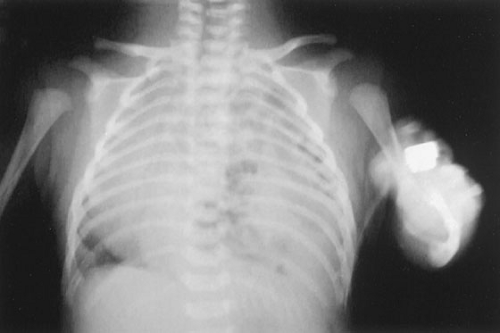 FIGURE 58-4. A chest radiograph of a newborn with a left-sided CDH. The heart can be seen shifted to the right and there are multiple air filled bowel loops in the left chest. |
Medical Therapy
Experimental models of CDH have suggested that the lungs of affected animals are immature in a number of ways. One observation that has led clinicians to attempt therapeutic intervention is that both rodent and sheep in models of CDH appear to be surfactant deficient (19,20). Suen and colleagues showed decreased levels of disaturated phosphatidylcholine, total lung DNA, and total lung protein, all indicative of lung immaturity. In animals with CDH, antenatal glucocorticoid administration has been shown to increase heart size, increase surfactant protein synthesis and endothelin receptor levels, increase total lung protein synthesis, and upregulate peptide growth factor gene expression (21,22,23).
The use of glucocorticoids in humans followed these reports in the hope that the fetus would increase intrinsic surfactant production. Despite the clear benefits of antenatal corticosteroids in animal models, there are many known potential adverse effects in humans (24). Because corticosteroids are widely used in obstetric practice for fetuses at risk of preterm delivery, their use in mothers carrying a fetus with CDH has been embraced in a number of institutions. However, there are at present no clinical data that show clear benefit from the use of antenatal corticosteroids in humans with regard to CDH. A case series of three surviving patients using a very prolonged course of antenatal steroids has been published (25). These data are anecdotal, and the use of prenatal corticosteroids to improve outcome in fetuses with CDH should be considered unproven at present. Other agents, such as thyrotropin-releasing hormone, have been shown to influence lung development in animal models of CDH (26). None have been studied clinically, and there are no accepted indications for their use in patients.
Fetal Surgical Therapy
Reports showing a poor outcome for fetuses diagnosed with CDH, as well as the high infant mortality seen in the 1980s, prompted a search for an approach to correct the malformation in utero. The concept of correction during a period of in utero support at a point where subsequent lung development could occur, was and is theoretically attractive. The first reported successful repair of a fetal CDH was by Harrison and colleagues in 1990 (27). Although in utero repair is possible for fetuses without a herniated liver, there has been no significant difference in mortality between an in utero open repair group and a conventionally treated group. When the liver was herniated into the chest, in utero operative reduction resulted in acute obstruction of umbilical venous flow and fetal death. Interest in the open antenatal surgical approach to CDH waned after a series of poor outcomes, and this approach was abandoned (28). A major concern that has been noted previously, is difficulty in correctly stratifying high- versus low-risk fetuses (29). This is recently reviewed (30).
With the observation that spontaneously occurring laryngeal atresia was associated with lung hypertrophy, Wilson and colleagues devised a series of studies that showed that tracheal ligation in experimental fetal animals resulted in larger and more mature lungs at birth (31). In an attempt to use this information in fetal intervention relevant to CDH, a technique known as PLUG (plug the lung until it grows) was developed (32). A series of reports followed, with varying modifications on how to safely and transiently occlude the trachea without significant long-term damage (33). Although human CDH survivors have been reported using this approach, concerns that tracheal occlusion may delay pulmonary maturation, a lack of controlled trials, and the risks of preterm labor continue to limit fetal intervention. A federally funded randomized trial of tracheal occlusion was halted early as outcomes in the fetal treatment group were not superior to conventional therapy following delivery. Despite a large literature on the topic, fetal intervention (both medical and surgical) has not been proven to benefit fetuses with CDH and, currently, there are no indications for fetal intervention in patients with CDH.
Postnatal Management
Prenatal diagnosis of CDH enables the medical team to be present at delivery for those infants and, most important, allows prenatal parental counseling. Known CDH patients should clearly be delivered at an institution that has the necessary equipment and personnel to offer contemporary management of most of the problem. Infants with respiratory distress should be treated initially with endotracheal intubation. Keeping inspiratory pressures low is a priority because even a short duration of high-pressure ventilation can cause lung injury (34). Management options for these patients encompass a number of different modalities, and there is no consensus on a single best approach. Many questions exist regarding the benefit and appropriateness of new techniques and therapies. Some of the issues in the management of CDH include best ventilator strategies, high-frequency oscillatory ventilation (HFOV), inhaled NO, exogenous surfactant, and ECMO.
Ventilator Strategy
Mechanical ventilation is the initial therapy for infants with respiratory failure from CDH. With the advent of neonatal mechanical ventilation in the 1960s, many CDH infants with previously fatal respiratory failure were surviving long enough to undergo surgical repair. As a result, the mortality of CDH climbed during that time due to an increase in both the number of patients undergoing operation and the severity of illness in those patients. At the onset, the goal of mechanical ventilation was adequate oxygen delivery. As understanding of perinatal physiology grew, the critical importance of pulmonary hypertension and right-to-left ductal shunting was appreciated. Boix-Ochoa first reported that differences in pre- and postductal pH and PaCO2 could be used to differentiate between survivors and non-survivors (35). Rudolf and Yuan then showed that the neonatal pulmonary vascular system was sensitive to changes in PaCO2 and pH. Drummond and others demonstrated that by increasing pH and decreasing PaCO2, ductal shunting could be reversed (36). Despite limited number of patients in this report, this resulted in the era where hyperventilation was widely used as the primary ventilator strategy for CDH patients. However, the high ventilator rates and inspiratory pressures caused significant barotrauma. This strategy persisted until Wung and others demonstrated that much of the mortality in CDH infants was, in fact, due to ventilator-induced lung injury (37). During this period, it was shown that lung injury and mortality were reduced in neonates who met ECMO criteria and thus had a period of “lung rest” while supported on ELMO. From the 1990s and onward, ventilation strategy in most centers has focused on minimizing barotrauma by allowing spontaneous ventilation with minimal set respiratory rates, the use of pressure-limited ventilation, tolerance of high PaCO2, minimal sedation, and avoidance of paralysis. The goal of ventilator support is to afford adequate hemoglobin saturation with oxygen and to maintain an acceptable arterial pH. Using this concept of permissive hypercapnia, several authors have reported survival rates approaching 90% (38,39,40).
High-Frequency Oscillation
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


