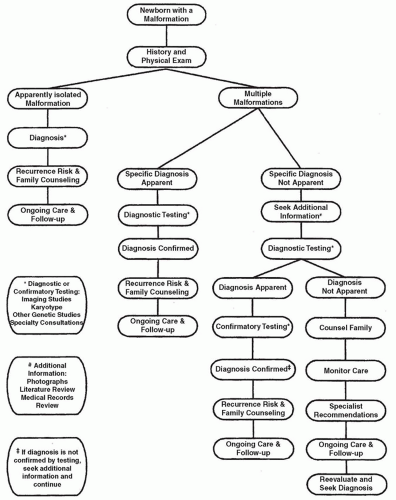
Over the past 60 years, a number of technologies have improved our understanding of the genetic bases for congenital malformation. At the present time, array comparative genomic hybridization (aCGH), more commonly known as CMA analysis, is recommended as a first-tier genomic test for most newborns with multiple congenital anomalies. Traditional karyotyping still offers important
advantages in some clinical scenarios, especially for confirmation of a clinically obvious trisomy, but has more of an adjunctive role. Fluorescent in situ hybridization (FISH) testing using targeted probes, such as for the DiGeorge Critical Region on chromosome 22q11.2; biochemical assays; single-gene testing; and multigene testing using massively parallel sequencing technologies are the investigations of choice in selected circumstances.
Chromosome Analysis
Karyotyping in the cytogenetics laboratory requires cell culture and preparation of numerous chromosome “spreads,” which are then analyzed to determine if all 46 chromosomes are present and to identify aberrant patterns of banding. Preparation, analysis, and reporting usually require at least 3 days. Forty-six chromosomes are present in most normal human cells. Formation of ova and sperm, however, is a surprisingly error-prone process: About one-half of all fertilizations result in aneuploidy or abnormal chromosomal number or structure, with subsequent reproductive loss. Some of this prenatal loss occurs late enough to be recognized as a miscarriage or spontaneous abortion, but most wastage is occult. Ninety-eight percent of these chromosomal defects are lethal (
15). In clinically recognized pregnancies, numerical chromosome abnormalities occur in 3% to 4% (
16).
An abnormal karyotype occurs in about 1 of 150 live born infants (
16,
17). Among chromosomally abnormal neonates, one-third have an extra sex chromosome with mild or no phenotypic manifestations in the newborn period; one-fourth have trisomy of an autosome, such as trisomy 21 or trisomy 18; and 40% have a variation of chromosomal structure, such as a deleted or duplicated segment or a translocation. Of the latter, most (79%) are balanced and generally do not cause birth defects. Approximately 10% of infants who die in the perinatal period secondary to multiple congenital malformations have abnormal cytogenetic studies (
15). Isolated malformations are very infrequently caused by a cytogenetically visible defect. On the other hand, neonates with multiple major malformations or a single major malformation accompanied by several minor malformations, especially when accompanied by a generalized dysmorphic appearance, intrauterine growth restriction, or an abnormal neurologic exam, are more likely to have an abnormal karyotype.
Fluorescent In Situ Hybridization
Many syndromic conditions are caused by submicroscopic chromosomal deletions that are not visible by routine karyotyping. Molecular (DNA) probes that hybridize at these loci can be complexed with a fluorescent marker to become powerful tools for detecting otherwise occult chromosomal deletions and other structural rearrangements. This technique, termed FISH, can be adapted for virtually any location or segment of the genome, including whole chromosomes and known hotspots, such as the “critical regions” for DiGeorge syndrome, Williams syndrome, and Prader-Willi syndrome. FISH technology is adept at delineating the nature of both straightforward and complex cytogenetic rearrangements, such as reciprocal translocations and insertions. Parental FISH studies are commonly recommended for neonates with an unbalanced translocation, in which there is both a chromosomal deletion and a duplication. Not uncommonly, one parent may harbor a balanced translocation, the understanding of which is critical for genetic counseling about recurrence risk. As a first-line investigation, however, FISH studies are primarily useful to confirm a submicroscopic deletion, which the clinician strongly suspects on the basis of clinical evidence. “Long-shot” suspicions are better addressed by screening the genome with other techniques, such as CMA analysis.
Chromosomal Microarray Analysis
Comparative genomic hybridization—the comparison of one genome to another using DNA hybridization—emerged in the 1990s as a useful research technique for understanding the complex cytogenetic rearrangements of malignancies. This process involved a pairwise matching of a normal genome with another suspected to be abnormal. Monosomy and trisomy for both large and small regions of the patient’s—or the tumor’s—genome could be easily identified. In the early 2000s, in order to create a more nimble clinical version, several investigators devised an adaptation in which only selected regions of the genome were compared. These regions, organized in arrays, were created using a few thousand bacterial artificial chromosomes (BACs) and were, by today’s standards, fairly large and few in number. However, the BAC aCGH was an immediate boon for clinical diagnosis because the analytic process was fast, could be automated, and improved the genomic resolution by several orders of magnitude. Soon known as CMA analysis, this process could easily and simultaneously investigate thousands of submicroscopic regions of the genome, detecting both deletions and duplications and defining breakpoints with impressive accuracy. CMA technical platforms quickly evolved to replace the relatively large BACs with smaller DNA probes (oligonucleotides) and polymorphic single nucleotides (SNPs), resulting in an exponential increase in resolution. Carefully crafted to uniformly cover the overall structure of each chromosome, CMAs have also been constructed to examine the fine structure of known genomic deletion/duplication hotspots and evaluate the internal integrity of individual genes. To understand the extent to which the diagnostic paradigm has been shifted, consider that, in 1990, the best option for genomic testing was the prometaphase karyotype, which has a resolution of 600 to 850 bands. CMAs now routinely use combinations of oligonucleotides and SNPs that exceed two and one half million, a nearly 3,000% improvement in resolution (
18).
CMA results are reported using nomenclature developed by the International Cytogenetics Standards Association. A normal array is diploid at all locations and will be described as “arr x2.” A deletion is designated by first identifying the chromosome arm and band, followed by a parenthetical description of the DNA base numbers at the start and the end of the deletion. “x1” would mean only one copy is present, that is, a deletion or monosomy. “x3” indicates three copies—a duplication or trisomic section. Many laboratories further describe the deletion or duplication in terms of size—either kilobases or megabases. Deletions and duplications are termed “copy number variants” (CNVs) and may be pathogenic, benign, or of uncertain/unknown significance. CNVs are quite common in the general population and most often do cause disease. Small CNVs in particular are ubiquitous and most likely benign, though a deletion of even a few base pairs at a critical location in an important gene can have significant consequences. As a general rule, deletions that are smaller than 0.5 Mb are likely to be benign, and those that exceed 1.0 Mb are more likely to have a deleterious effect. The genetic laboratory evaluates all CNVs to render an opinion about probable significance and includes a summary of their opinion in the CMA report. In addition to the size and location of the deletion, the laboratory determines which specific genes might be deleted or duplicated and reviews the medical literature to see if alterations of these genes have been reported to cause human disease (
19). The laboratory’s medical director or liaison, often a genetic counselor, can provide superior analysis of CMA results when they have a clear understanding of the patient’s phenotype. Conversely, the neonatal intensive care unit (NICU) provider usually gains significant insight into the meaning of a CMA result after a discussion with the laboratory.
To clarify abnormal CMA results, parental genetic testing is often useful. Occasionally, this may require CMA testing of each parent, but more often, parents are evaluated with FISH analysis using a DNA probe that hybridizes to the child’s CNV. Parental FISH testing may reveal that the parent has the same CNV and in fact may have a syndromic diagnosis. For instance, CMA for a child with tetralogy of Fallot may reveal the chromosome 22q11.2 deletion of DiGeorge syndrome, and the mother, who has a repaired cleft
palate, may have the same deletion detected via FISH. FISH testing can also reveal that a parent carries a balanced translocation, an important discovery for the purpose of recurrence risk counseling, though less germane for the management of the newborn.
Variants of uncertain significance, often abbreviated as VUS or VOUS, are reported in 5% to 10% of CMA results. To clarify these types of results, the laboratory often suggests that parental FISH studies be obtained. If a parent is phenotypically normal and shares the same CNV, then the chances increase that this is a benign variant. Conversely, if the CNV has occurred only in the child, that is, is a de novo event, then the variant is more likely deleterious. Another strategy is to reanalyze the VUS at a later time, when additional observations are available to the laboratory through their own internal experience, shared databases, and the medical literature. Upon request, most laboratories will gladly attempt to reclassify VUS.
CMA analysis is often selected as a diagnostic tool to seek small, clinically significant CNVs, but it is easily capable of detecting larger genomic aberrations, such as trisomy 21 and other conditions that would be visible using standard chromosome testing. If Down syndrome, trisomy 13, trisomy 18, or another aneuploidy is strongly suspected on clinical grounds, then karyotyping, not CMA, is the diagnostic study of choice. If a microdeletion syndrome, such as Williams syndrome or Miller-Dieker syndrome, is strongly suspected, site-specific FISH studies are reasonable first tests. For neonates with multiple malformations or dysmorphic features that are not recognized as a specific diagnosis, CMA is the preferred, first-tier investigation. A number of studies have demonstrated that CMA, compared to cytogenetic studies of the genome, has a significantly better yield (
20,
21). One may expect that about 20% of newborns with more than one major malformation will have a confirmed, deleterious CNV using CMA. In addition, prenatal CMA also has significant advantages over standard karyotyping, and these results may be available via analysis of chorionic villus tissue or amniotic fluid. For pregnancies with a structural anomaly on ultrasonography and a normal prenatal karyotype, 6% will have a pathogenic CNV on prenatal CMA (
22).
CMA has some limitations and some unique features. CMA is not as fast as standard karyotyping, with results available in 1 or 2 weeks, as opposed to several days for chromosomes and FISH. CMA is incapable of determining the spatial relationships of the oligonucleotides and SNPs used in the array. Consequently, it will not detect balanced translocations or chromosomal inversions. In those instances, the break points may on rare occasions disrupt the coding sequence or internal structure of a gene, resulting in haploinsufficiency and possible phenotypic consequences. CMA will not detect triploidy. CMA tests that use SNP technology are also capable of detecting regions of homozygosity (ROH). Small ROH may reflect uniparental disomy (UPD); extensive and multiple large ROH often indicate consanguinity. Identification of possible consanguinity raises a number of ethical, social, and legal issues. In some instances, parents are unaware of their biologic relationship, but occasionally, ROH is extensive enough to suggest that the newborn is the product of an incestuous relationship. In each case, there is increased risk for homozygous mutations, resulting in a recessive condition. UPD may cause malformations via abnormal imprinting. If at all possible, a frank discussion with parents—prior to testing—about the possibility that SNP-CMA testing may reveal consanguinity would be prudent and is increasingly recommended (
23).
Single-Gene and Multigene Analysis
Newborns recognized or suspected as having a specific genetic condition may benefit from molecular confirmation using targeted analysis or sequencing of a single or several genes. For instance, for a child suspected to have achondroplasia, a DNA nucleotide change of the fibroblast growth factor receptor-3 (FGFR3) gene at position 1138 accounts for 99% of cases, so targeted testing at this locus is most logical. More often, however, single-gene analysis requires sequencing—a base pair by base pair analysis of the coding portion of either the entire gene or selected exons of the gene—and, in many instances, deletion/duplication analysis to detect small or large intragenic or whole gene deletions and duplications. The genetic sequencing techniques developed by the Nobel laureate Frederick Sanger (Sanger sequencing) are the industry standard. Detection of deletions and duplications requires other technologies, such as targeted genomic hybridization or multiplex ligationdependent probe amplification.
For some diagnoses, mutations in multiple genes may cause the same phenotype. Examples include Noonan syndrome, holoprosencephaly, and Bardet-Biedl syndrome. Genetic testing in these scenarios may be conducted stepwise: testing one gene at a time, beginning with the most common or likely candidate gene. However, this tactic is both expensive and time consuming. Simultaneous testing of a panel of genes using Sanger sequencing has become more common and does speed up the analytic process.
A new technology—massively parallel sequencing—has emerged as a more cost-effective method to rapidly and simultaneously sequence large numbers of genes. Also known as next-generation sequencing (NGS), this technology can generate sequence data for the great majority of the human genome—nearly three billion base pairs of DNA (
24). Whole-genome sequencing, though highly promising and likely to become a clinical tool within a few years, is currently a research tool (
25). However, nearly any subset of the whole genome can be selected for analysis, and a popular current strategy is whole-exome sequencing (WES), discussed below. Selection of other subsets is both straightforward and practical, and NGS panels are readily available from multiple laboratory vendors. These panels can range in size from less than a dozen genes to several hundred genes and may specifically target a single diagnosis, such as Noonan syndrome, or a closely related group of diagnoses, such as Marfan syndrome, Loeys-Dietz syndrome, and related conditions. NGS panels offered by various laboratory vendors are themed to evaluate mitochondrial disease, cardiac conduction defects, epilepsy, autism, intellectual disability, and more.
The most comprehensive NGS panel clinically available at this writing is WES, which evaluates approximately 90% of all exons— the expressed or coding sequences of genes, which constitute about 2% of the entire genome. Among children with undiagnosed neurologic disease, WES can establish a firm diagnosis in 25% to 50% (
26), an impressive yield that suggests to some observers that WES might be an effective first-tier investigation that would avoid the expense, time, and frustration of the diagnostic odyssey (
27). However, there are a number of logistic, financial, and ethical issues that make WES less practical for use in the newborn population. At this time, the turnaround time is between 4 and 6 months, and the cost is between $7,000 and $12,000. Many payers consider any test that uses NGS experimental and investigational, and extensive pretest genetic counseling by a genetic counselor or other genetics professional is generally recommended in order to educate parents about the nature of the test, the expected yield, the potential to reveal nonpaternity and consanguinity, the possibility that a diagnosis will not be established, and the many expected variants of uncertain significance. In addition, parents must be informed that WES is capable of detecting deleterious mutations in genes unrelated to the clinical situation for which they are seeking answers, such as adult-onset susceptibility to breast and ovarian cancer, and will also reliably detect if their child is a carrier for recessive diseases. The degree to which families may exercise control over learning of these incidental findings has been a topic of keen ethical debate. Laboratories that offer WES will generally allow parents to choose to not receive those incidental results, in accordance with revised practice guidelines of the American College of Medical Genetics (
28,
29,
30).