Cleft Lip and Cleft Palate
Alex A. Kane
Plastic and Reconstructive Surgery, Washington University School of Medicine, St. Louis Children’s Hospital, St. Louis Missouri 63110.
INTRODUCTION
Cleft lip and palate are the most common congenital craniofacial anomalies in humans, affecting approximately 1 in 700 live births. Clefts of the lip and palate occur in a spectrum of anatomic severity and are often found in association with anomalies of other organ systems. Both cleft lip and cleft palate can occur in isolation. Estimates vary, but roughly one-half (46%) of affected individuals have both cleft lip and palate, whereas isolated cleft lip affects one-fifth (21%) and isolated cleft palate occurs in one-third (33%) of cases (1).
Depending on cleft severity, the child with cleft lip and palate often requires a series of surgical interventions that are staged throughout infancy and adolescence. The order and selection of procedures are influenced by developmental and functional concerns. Usually, the cleft lip is repaired first and the cleft palate second, and both are normally performed within the first year of life. Secondary management for velopharyngeal (VP) dysfunction is undertaken if indicated in early adolescence. Bone grafting of the alveolar ridge defect is performed during the mixed-dentition stage at age 8 to 11 years. During adolescence, nasal reconstructive surgery, including augmentative tip rhinoplasty, or septoplasty is performed as indicated. Orthognathic jaw surgery to correct growth discrepancies between the upper and lower jaws is usually last in the treatment sequence, performed after dentoskeletal maturity in the teen years. The specific operations, maneuvers, and sequencing performed to address these problems vary considerably between cleft surgeons, according to training, experience, and preference.
The Cleft Team Concept
The existence of a facial cleft in a child calls for a long-term treatment plan. Both therapy and rehabilitation begin soon after birth, and most patients continue therapy of some sort until they reach their late teens or early twenties. Current standards of cleft care include comprehensive multidisciplinary management by a cleft palate team. The concept of an organized team evolved to address the fragmented care provided by independent specialists in previous years. The fundamental principle driving this approach involves sharing information and cultivating a global perspective about a given patient’s needs.
Children born with clefts face multiple and complex health problems. Experience has shown that these complex issues can best be managed by an interdisciplinary team of specialists. Depending on the child’s individual needs (2,3,4), these specialists may include an audiologist, a craniofacial surgeon (who might be trained in plastic surgery, otolaryngology, or oral and maxillofacial surgery), pediatric dentist, prosthodontist, orthodontist, geneticist, cleft nurse specializing in cleft feeding and growth issues, pediatrician, psychologist or other mental health specialist, and speech and language pathologist. When these specialists work together and with the family, treatment goals can be individualized for each child, and parents and health care providers can make the best choices for treatment by consulting with each other. Because growth is a significant factor in the ultimate outcome of treatment, the child must be assessed thoroughly and regularly by the team until young adulthood (5).
Feeding the Infant with Cleft Palate
Infants whose cleft involves the palate usually cannot generate enough suction to draw milk from breast or bottle. This is because an intact palate is necessary to generate the negative pressure necessary for an effective suckling mechanism. A variety of cleft palate bottles exist, which work by application of positive pressure, compensating for the lack of negative pressure generated by the child. These are used with various nipples to deliver formula or pumped breast milk to these infants. The techniques employed to feed infants with clefts require practice and patience for the
parent, who are assisted by nursing professionals specialized in this area. Infants with a cleft lip and palate require frequent burping because they swallow more air, due to the application of positive pressure while feeding. Babies with isolated cleft lip can often nurse from the breast or from normal bottles if care is taken to position the child’s lip to allow for an effective seal to develop.
parent, who are assisted by nursing professionals specialized in this area. Infants with a cleft lip and palate require frequent burping because they swallow more air, due to the application of positive pressure while feeding. Babies with isolated cleft lip can often nurse from the breast or from normal bottles if care is taken to position the child’s lip to allow for an effective seal to develop.
Embryology: Normal and Abnormal Development of the Lip and Palate
It is generally accepted that facial tissues, including the lip and palate, arise from neural crest cells (6). These cells migrate from their original positions at the margins of the neural fold, providing the mesenchyme for the paired maxillary and mandibular processes (which are in turn derivatives of the first and second branchial arches), and the central frontonasal process. Bilateral nasal pits develop at the inferolateral aspects on either side of the frontonasal prominence. The tissues on the sides of the nasal pits become the medial and lateral nasal processes. Normally, the medial nasal prominences fuse with the maxillary processes below the nasal pit to form the upper lip. Failure of fusion results in cleft lip. The frontonasal process does not contribute to the lip, but develops into the primary palate, which is the portion of the palate containing the alveolar bone between the canine teeth, extending posteriorly to the incisor foramen (Fig. 54-1).
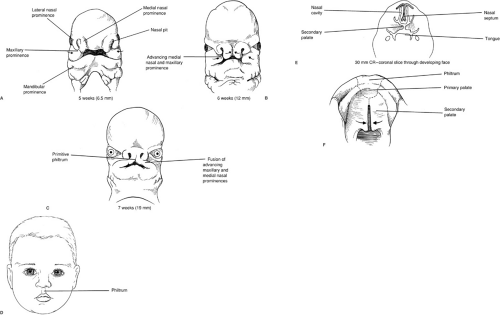 FIGURE 54-1. Embryology of cleft lip and cleft palate. |
The secondary palate, containing the alveolus, hard palate, and soft palate posterior to the primary palate, arises as bilateral outgrowths from the maxillary processes. These shelves initially grow vertically down the side of the tongue, then elevate at a precise time to a horizontal position above the dorsum of the tongue, and fuse with each other to form an intact palate. Interference with shelf elevation is the putative mechanism by which most cases of human cleft palate arise. Cleft palate may also result from disturbances in shelf growth, defective shelf fusion, failure of medial edge cell death, postfusion rupture, and failure of mesenchymal consolidation and differentiation.
Agents that can negatively affect the growth and development of the primary palate (i.e., a cleft extending ventrally from the incisive foramen) include Dilantin (phenytoin), alcohol, hypoxia, retinoids (member of the vitamin A family), and possibly dietary factors such as folate vitamin deficiency.
Genetics of Clefting
The genetics of orofacial clefting are only partially understood, but of great importance in counseling affected families. It is generally believed that isolated cleft palate (CP) is a distinct genetic entity from unilateral cleft lip with or without cleft palate (CL/P) (2,3,4). This conclusion has followed from both epidemiologic studies and the fact that embryologic events leading to CL/P and CP occur at
somewhat different times (3 to 7 weeks vs. 5 to 12 weeks). It has long been assumed that both genetic and environmental (epigenetic) factors play important roles in the etiopathology of clefts, and this is supported by the varying incidence of clefting with ethnicity, geographic location, and socioeconomic conditions (7,8,9,10). Twin studies have clearly demonstrated a genetic basis for CL/P, with a 43% pairwise concordance rate in monozygotic twins versus a 5% concordance in dizygotic twins (2,11,12).
somewhat different times (3 to 7 weeks vs. 5 to 12 weeks). It has long been assumed that both genetic and environmental (epigenetic) factors play important roles in the etiopathology of clefts, and this is supported by the varying incidence of clefting with ethnicity, geographic location, and socioeconomic conditions (7,8,9,10). Twin studies have clearly demonstrated a genetic basis for CL/P, with a 43% pairwise concordance rate in monozygotic twins versus a 5% concordance in dizygotic twins (2,11,12).
The incidence of CL/P in white newborns is approximately 1 in 700, with isolated cleft palate occurring in about 0.5 in 1,000. The incidence of CP alone is significantly less than CL/P, occurring one-third to one-half as often (7). Whereas there are more than 250 syndromes associated with orofacial clefting (8), it is generally considered that the great majority of cases occur as an isolated abnormality, so-called “nonsyndromic” CL/P (7). Given the incomplete understanding of the genetics of orofacial clefting and imprecision in employing “nonsyndromic” versus “syndromic” nomenclature, estimates vary with regard to the frequency with which other malformations occur in children with clefts. In a large review of their center’s experience, Rollnick and Pruzansky (13) identified other malformations in 35% of CL/P patients and 54% of CP patients. CL/P has an unequal gender distribution, favoring boys over girls, whereas this relationship is reversed in CP (14). CL/P affects the left side more often (15,16).
Unaffected (i.e., noncleft) parents who have one child with CL/P have an estimated recurrence risk of 4%, which rises to 9% with two affected children. If one parent is affected, the risk of having a child with CL/P is also 4%, increasing to 17% if there is already both an affected parent and affected child (17). As the degree of familial relationship increases, recurrence risk decreases, with first-, second-, and third-degree relatives having 4%, 0.7%, and 0.3% risk, respectively (9). Recurrence risk increases with the severity of the cleft (18).
The most appropriate genetic model for the inheritance pattern of nonsyndromic CL/P is a matter of considerable debate. Classically, Fogh-Anderson proposed the idea that CL/P was transmitted by a gene of variable penetrance that could act dominantly or recessively, depending on the individual.(2,9). Multifactorial and multifactorial/threshold models have been widely advanced (19) and have predominated, whereas others believe there is little evidence to support the concept of transmission as a discontinuous threshold trait (7). Since the mid-1990s, the prevailing method of genetic analysis of CL/P has been the use of allelic association studies, whereby “candidate” genes are selected on the basis of functional properties, expression pattern, chromosomal location, or mouse homologues (19). Numerous studies of this type have found significant association to the transforming growth factor alpha (TGFA) (20), transforming growth factor beta 3 (TGFB3) (21), retinoic acid receptor alpha (RARA) (22), the homeobox gene MSX-1 (21), and the BCL3 protooncogene (23) loci. Although such studies are useful, their results must be interpreted with caution because it is not clear whether the ever-increasing number of implicated loci are truly representative of a large number of genes involved in the etiology of CL/P, or that this type of analysis has the ability to produce false-positives (9,19).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


