Central Nervous System
Herbert Edgar Fuchs
Department of Surgery, Duke University, Pediatric Neurosurgical Services, Department of Surgery, Duke University Medical Center, Durham, North Carolina 27710.
The surgeon who works with the developing central nervous system (CNS) must have a thorough knowledge of the embryology of the nervous system to understand and effectively treat the vast array of complex malformations, tumors, and developmental disorders that can occur. This chapter reviews the development of the CNS and the spectrum of nontraumatic disorders that can affect the developing nervous system.
NORMAL DEVELOPMENT
Human embryonic development is divided into 23 morphologic stages, each of which lasts 2 to 3 days (1). These stages encompass the period from fertilization to about 60 days’ gestation and have also been correlated with those in animals, particularly rodents and birds. Although there are some important interspecies differences in CNS development, animal model systems have provided great insight into the processes involved in human CNS development, and therefore, are included in this discussion (2).
Neurulation
The neural tube forms by a process termed neurulation, during embryonic stages 8 to 12, at 18 to 27 days’ gestation. During this phase, the most severe open forms of cranial and spinal dysraphism occur. In the later stages of embryonic development, the caudal neural tube is formed by canalization and regression. During this period, the occult forms of dysraphism can develop. The notochord induces the overlying ectoderm to form the neural plate, which is contiguous laterally with the superficial ectoderm, by the end of the third week of gestation. During the next several days, the lateral portions of the neural plate begin to elevate and to form the neural folds as the midline portion becomes the neural groove (Fig. 108-1). As this process continues, the neural folds fuse in the midline to form the neural tube, beginning in the cervical region and proceeding both cranially and caudally. The anterior and posterior neuropores close at about 23 and 25 days’ gestation, respectively. Immediately after fusion of the neural folds, the superficial ectoderm fuses in the midline and then separates from the neural ectoderm (Fig. 108-1). Mesenchymal cells then migrate between the skin and the neural tube, ultimately to form the meninges, neural arches, and paraspinal muscles.
Caudal Regression
After neurulation is complete, by day 25, the distal spinal cord begins to form as the caudal end of the neural tube blends into the caudal cell mass, a large mass of undifferentiated cells. The caudal cell mass eventually gives rise to components of the nervous, urogenital, and digestive systems. This accounts for the common joint occurrence of distal vertebral, neural, anorectal, and urogenital anomalies. Within the caudal cell mass, small vacuoles form, coalesce, and eventually connect with the central canal of the spinal cord, in the process known as canalization (Fig. 108-2). The surrounding cells differentiate toward glial cells, elongating the spinal cord well into the tail fold of the embryo. The most cephalic portion of this distal spinal cord forms the tip of the conus medullaris. The distal spinal cord then begins the process of involution or retrogressive differentiation, leaving a remnant of piaarachnoid, the
filum terminale. From this point, the spinal cord and developing vertebral column elongate, with the vertebral column growing faster than the spinal cord. Thus, the conus medullaris appears to ascend from its initial position in the coccyx, to lie at the L2–3 interspace by birth. By 3 months postpartum, the tip of the conus medullaris is nearly at the adult level of the L1–2 interspace (3).
filum terminale. From this point, the spinal cord and developing vertebral column elongate, with the vertebral column growing faster than the spinal cord. Thus, the conus medullaris appears to ascend from its initial position in the coccyx, to lie at the L2–3 interspace by birth. By 3 months postpartum, the tip of the conus medullaris is nearly at the adult level of the L1–2 interspace (3).
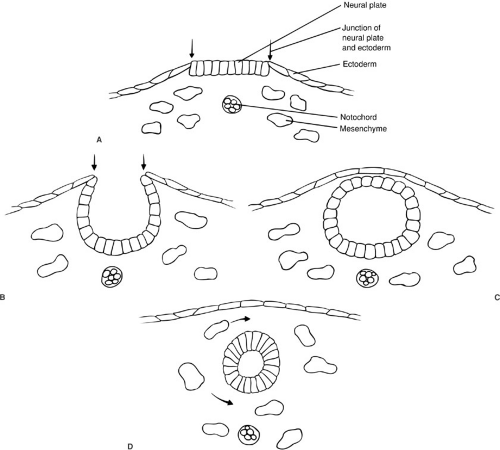 FIGURE 108-1. Neurulation. (A) Cross section of an embryo, showing the neural plate. (B) Formation of the neural folds (arrows). (C) Formation of the neural tube. (D) Separation of the neural tube from the superficial ectoderm by mesenchymal cells. |
Formation of the Brain
By embryonic stage 10 (days 22 and 23), the cephalic neural folds distinguish the future brain from the future spinal cord. Two constrictions divide the developing brain into three regions: the prosencephalon (forebrain), mesencephalon (midbrain), and rhombencephalon (hindbrain). By 35 days, the prosencephalon has divided into the telencephalon (future cerebral hemispheres) and the diencephalon, whereas the rhombencephalon has formed the metencephalon (future pons) and the myelencephalon (future medulla oblongata). Both cerebral hemispheres contain a cavity, the lateral ventricle. Glial and neural precursor cells begin to form around this cavity and migrate radially outward toward the surface of the cerebral hemisphere. The lateral, third, and fourth ventricular
cavities are contiguous with the central canal of the spinal cord at this stage; later development obliterates the site of communication at the obex in the fourth ventricle. The cerebellum forms from a complex series of buckling and folding in the rhombencephalon, initially lying within the fourth ventricle, and eventually coming to lie dorsal to the fourth ventricle.
cavities are contiguous with the central canal of the spinal cord at this stage; later development obliterates the site of communication at the obex in the fourth ventricle. The cerebellum forms from a complex series of buckling and folding in the rhombencephalon, initially lying within the fourth ventricle, and eventually coming to lie dorsal to the fourth ventricle.
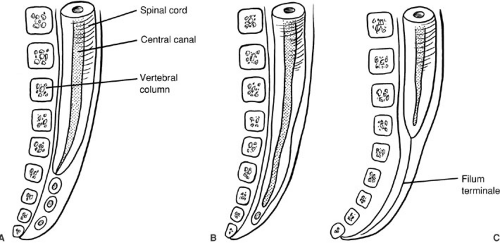 FIGURE 108-2. Formation of the caudal spinal cord and filum terminale. (A) Vacuolization. (B) Vacuoles coalesce; canalization occurs. (C) Differential growth and retrogressive differentiation cause the conus medullaris to rise and the filum terminale to lengthen. |
The choroid plexus first appears in the fourth ventricle at days 43 to 44, in the lateral ventricles at days 45 to 46, and in the third ventricle at days 48 to 49. The caudal roof of the fourth ventricle perforates to form the foramen of Magendie at days 47 to 48; at the same time, the choroid plexus begins to produce cerebrospinal fluid (CSF). The CSF loosens the meshwork of mesenchymal cells surrounding the brain to form the subarachnoid space, beginning at the cisterna magna, and spreading over the cerebral hemispheres and the spinal cord.
Formation of the Skull and Vertebral Column
The skull is formed in two sections: The skull base is formed by endochondral ossification, and the cranial vault is formed as membranous bone. The first chondrification begins during stage 17 (days 40 to 44). The vertebral column originates from the medial portion of the somites in three stages: membrane formation (days 22 to 39), cartilage formation (days 40 to 64), and bone formation, which extends through the fetal portion until well after birth. The notochord appears to direct formation of the vertebral bodies, and the neural tube directs formation of the posterior vertebral elements. Remnants of the notochord remain in the intervertebral disks and in the clivus of the skull.
CONGENITAL MALFORMATIONS
Congenital malformations of the CNS are best understood as derangements of the developmental processes discussed earlier (Table 108-1).
Derangements of Neurulation
Myelomeningocele is the most common derangement of neurulation, occurring in about 1 in 1,000 live births.
This incidence appears to be decreasing with emphasis on prenatal maternal care, particularly folate supplementation. It is estimated that up to 70% of myelomeningoceles could be prevented by women of childbearing age taking a multivitamin (4). In this malformation, a focal segment of the spinal cord fails to roll up and form a tube. Because the neural tube does not fuse, the cutaneous ectoderm does not come to cover the neural tube, but remains attached and lateral to the neural plate, leaving a cutaneous defect (Fig. 108-3). Mesenchymal cells are also prevented from their proper migration, and thus the laminal arches and muscles develop in an abnormal lateral position. The raw, exposed surface of the neural plate represents the interior of the spinal cord. The midline neural groove is also seen. Surrounding this neural placode is a thin layer of skin and arachnoid tissue, below which is the subarachnoid space. The nerve roots lie inferior to the neural placode, with the ventral roots lying medial to the dorsal roots. Because the neural placode is attached to the skin, it is tethered and relatively immobile. A number of CNS anomalies have been associated with myelomeningocele, including Chiari II malformation and hydrocephalus, which are discussed later in this chapter.
This incidence appears to be decreasing with emphasis on prenatal maternal care, particularly folate supplementation. It is estimated that up to 70% of myelomeningoceles could be prevented by women of childbearing age taking a multivitamin (4). In this malformation, a focal segment of the spinal cord fails to roll up and form a tube. Because the neural tube does not fuse, the cutaneous ectoderm does not come to cover the neural tube, but remains attached and lateral to the neural plate, leaving a cutaneous defect (Fig. 108-3). Mesenchymal cells are also prevented from their proper migration, and thus the laminal arches and muscles develop in an abnormal lateral position. The raw, exposed surface of the neural plate represents the interior of the spinal cord. The midline neural groove is also seen. Surrounding this neural placode is a thin layer of skin and arachnoid tissue, below which is the subarachnoid space. The nerve roots lie inferior to the neural placode, with the ventral roots lying medial to the dorsal roots. Because the neural placode is attached to the skin, it is tethered and relatively immobile. A number of CNS anomalies have been associated with myelomeningocele, including Chiari II malformation and hydrocephalus, which are discussed later in this chapter.
TABLE 108-1 Congenital Malformations of the Central Nervous System. | |
|---|---|
|
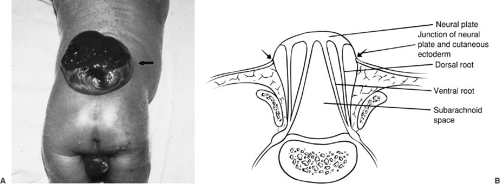 FIGURE 108-3. Myelomeningocele. (A) Myelomeningocele in a newborn (arrow). The anal sphincter is flaccid, and the lower-extremity musculature is poorly developed. (B) Schematic depiction of a myelomeningocele in cross-section. |
Lipomyelomeningocele is believed to form by focal premature dysjunction of neuroectoderm from cutaneous ectoderm, allowing access of mesenchymal cells to the dorsal surface of the unclosed neural tube (5). The mesenchymal cells then give rise to fat. The anatomy is therefore similar to myelomeningocele, with the exception that the cutaneous ectoderm is able to close over the neural tube. The lipoma may extend from the spinal cord and merge with the subcutaneous fat (Fig. 108-4).
In contrast to lipomyelomeningocele, dermal sinus tract results from incomplete dysjunction of the neural tube and cutaneous ectoderm. As the spinal cord becomes buried beneath the surface and elongates, the localized connection with the skin becomes an elongated tract (Fig. 108-5). The incidence of dermal sinus is highest in the lumbosacral region, the site of posterior neuropore closure, but this derangement can also occur in the cervicooccipital and nasal regions. It can extend to variable depths, ending in the subcutaneous tissues or the dura, or retaining its attachment to the spinal cord or brainstem. Dermoid or epidermoid tumors may be found along the course of the dermal sinus tract.
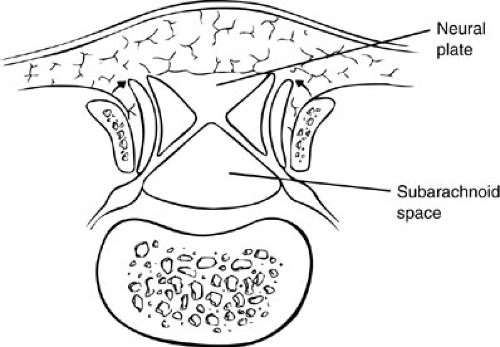 FIGURE 108-4. Cross-sectional drawing of a lipomyelomeningocele. |
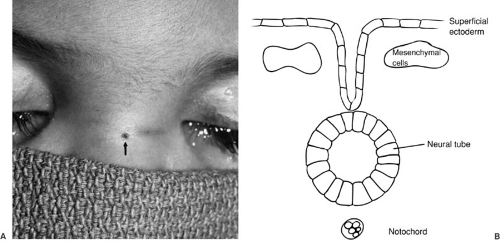 FIGURE 108-5. Dermal sinus tract. (A) Nasal dermal sinus tract (arrow). (B) Cross-sectional drawing of a dermal sinus tract. |
Derangements of Retrogressive Differentiation
The tight filum terminale or fatty filum terminale syndrome is a condition in which the spinal cord is tethered by an abnormally short, thickened filum terminale. The terminal myelocystocele is a complex malformation that is believed to be caused by dilation of the central canal in the caudal neural tube, forming a cyst. This cyst distends the arachnoid lining of the distal spinal cord, forming a meningocele. This anomaly is commonly associated with extrophy of the bladder.
Other Embryologic Derangements
Diastematomyelia is a condition in which the spinal cord is split in a sagittal plane into two hemicords that may or may not be symmetric. Diastematomyelia is most common in girls and is usually heralded by a hairy patch overlying the site of the cleft. The cause of this malformation has been a subject of great debate, but studies by Dias and Walker (6) and Pang and colleagues (7) support the concept that these malformations are due to disorders of gastrulation. Pang et al. classified these lesions as type I or II split cord malformation (SCM). Type I SCM is the classic diastematomyelia, with a bony septum lying between two hemicords, each lying in its own dural sac. The type II malformation consists of two hemicords in a single dural sac, with a thin sagittal fibrous septum. These malformations can be caused by splitting of the notocord, either by duplication or by persistence of the neurenteric canal, which effectively causes a localized split of the notocord. The development of mesenchymal elements between the two hemicords then determines the type of SCM formed. A thicker mesenchymal tract gives rise to the bony septum and meninges, whereas a thinner tract may only form a thin fibrous septum (Fig. 108-6).
A neurenteric cyst can also result from persistence of the neurenteric canal and is often associated with SCM. This rare lesion is most often seen as only a partial fistula and is most common in the cervicothoracic region of the spinal cord.
Meningoceles are believed to be due to postneurulation disorders involving cutaneous ectoderm and mesenchyme because the neural tube is normally formed beneath the cutaneous and mesenchymal defect, which contains CSF (Fig. 108-7).
The embryology of encephaloceles has been reviewed by Chapman and associates (8). Encephaloceles were originally believed to be caused by failure of closure of the anterior neuropore. These lesions, however, contain well-developed neural and mesenchymal structures, which cannot be the result of failure of neural tube closure. Therefore, these lesions are believed to be due to herniation of fully neurulated neural tissue through a mesenchymal defect (Fig. 108-8).
The Chiari II malformation is a complex disorder associated with myelomeningocele. This malformation consists of caudal displacement of the cerebellar vermis and tonsils into the cervical canal; elongation, kinking, and
caudal displacement of the lower brainstem into the cervical canal; and upward displacement of the superior cerebellum through a low-lying tentorial incisura, with a small posterior fossa (Fig. 108-9A). McLone and Knepper (9) proposed a unifying theory of pathogenesis of these malformations, in which the open neural placode of the myelomeningocele allows escape of CSF, which interferes with the normal distention of the ventricular system and development of the skull base. Incomplete distention of the fourth ventricle fails to stimulate growth of the skull base, resulting in a smaller posterior fossa that is unable to respond during the later phase of rapid cerebellar growth and development. This results in herniation of neural tissue from the posterior fossa and impairs flow of CSF, resulting in hydrocephalus. The Chiari I malformation, involving caudal displacement of the cerebellar tonsils with a normal posterior fossa, must have its embryologic origin at a later stage than the more severe Chiari II malformation, but the exact mechanisms responsible remain to be elucidated (Fig. 108-9B).
caudal displacement of the lower brainstem into the cervical canal; and upward displacement of the superior cerebellum through a low-lying tentorial incisura, with a small posterior fossa (Fig. 108-9A). McLone and Knepper (9) proposed a unifying theory of pathogenesis of these malformations, in which the open neural placode of the myelomeningocele allows escape of CSF, which interferes with the normal distention of the ventricular system and development of the skull base. Incomplete distention of the fourth ventricle fails to stimulate growth of the skull base, resulting in a smaller posterior fossa that is unable to respond during the later phase of rapid cerebellar growth and development. This results in herniation of neural tissue from the posterior fossa and impairs flow of CSF, resulting in hydrocephalus. The Chiari I malformation, involving caudal displacement of the cerebellar tonsils with a normal posterior fossa, must have its embryologic origin at a later stage than the more severe Chiari II malformation, but the exact mechanisms responsible remain to be elucidated (Fig. 108-9B).
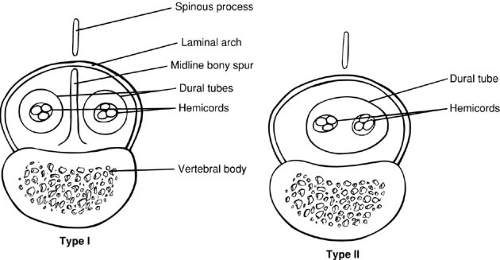 FIGURE 108-6. Split cord malformations depicted in cross section. |
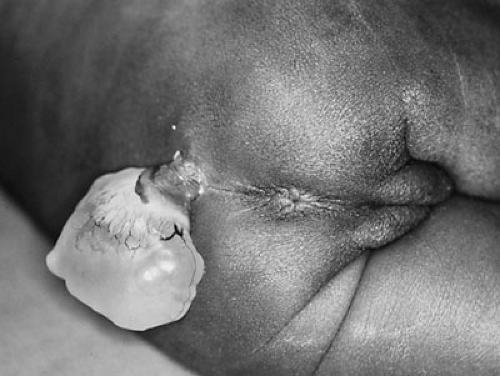 FIGURE 108-7. Infant with a sacral meningocele. The sac does not contain any neural elements. |
The Dandy-Walker malformation is a developmental abnormality in which the roof of the fourth ventricle fails to perforate to form the foramen of Magendie. The resultant cystic dilation of the fourth ventricle expands the posterior fossa, elevates the tentorium, and causes hydrocephalus due to obstruction of the aqueduct of Sylvius with concomitant hypoplasia of the cerebellar vermis (Fig. 108-10).
Arachnoid cysts are arachnoid-lined cavities filled with fluid similar in composition to CSF. They can create a disturbance in intracranial dynamics due to shift and displacement of surrounding structures, and they can cause intracranial hypertension. Their pathogenesis is unknown. Arachnoid cysts appear to form early in development and may communicate to varying degrees with the surrounding subarachnoid space. The most common locations for arachnoid cysts are the sylvian fissure, suprasellar, and posterior fossa.
Diagnosis and Management
Myelomeningocele
The diagnosis of myelomeningocele is obvious at birth. For initial management, the lesion is covered with sterile, saline-soaked dressings, and the patient is kept prone. After a general assessment, the location of the lesion is noted, and a neurologic examination is performed to assess sensorimotor function in the lower extremities. Hip flexion requires function of L1 to L3. Hip adduction requires L2 to L4 function. Hip abduction, hip extension,
and knee extension require L5 to S2 function. Plantarflexion requires function of sacral roots. Anal sphincter tone should be assessed because more than 90% of children with myelomeningocele have bowel and bladder dysfunction. Head circumference should be measured and the anterior fontanelle palpated to assess for hydrocephalus. After medical stabilization, and preferably within 24 hours of birth, the child should be taken to the operating room for closure of the myelomeningocele. This procedure involves meticulous anatomic reconstruction of the neural tube,
dural sac, and overlying cutaneous tissues, with great care taken not to further injure neural tissue. Postoperatively, the child is monitored for development of hydrocephalus, which is treated with a ventriculoperitoneal shunt, and nursed prone, while the closure heals. During this time period, further orthopedic and urologic workup can be completed, along with evaluation of brainstem function to assess the associated Chiari II malformation. The long-term outcomes for these children have been reported in depth by McLone and coworkers (10). The survival rate for these children followed for 8 to 12 years was 85%, with 62% having an IQ of 80 or higher. With the development of clean intermittent bladder catheterization, 85% of children with myelomeningocele can achieve social continence of urine. The development of improved leg braces has allowed children with motor levels of at least L3 function to be community ambulators. McLone’s studies have shown that children with myelomeningocele can be functional and that the natural history of this disease is not one of relentless deterioration, as some prior studies had suggested. Any neurologic deterioration seen in children with myelomeningocele should be promptly investigated and treated.
and knee extension require L5 to S2 function. Plantarflexion requires function of sacral roots. Anal sphincter tone should be assessed because more than 90% of children with myelomeningocele have bowel and bladder dysfunction. Head circumference should be measured and the anterior fontanelle palpated to assess for hydrocephalus. After medical stabilization, and preferably within 24 hours of birth, the child should be taken to the operating room for closure of the myelomeningocele. This procedure involves meticulous anatomic reconstruction of the neural tube,
dural sac, and overlying cutaneous tissues, with great care taken not to further injure neural tissue. Postoperatively, the child is monitored for development of hydrocephalus, which is treated with a ventriculoperitoneal shunt, and nursed prone, while the closure heals. During this time period, further orthopedic and urologic workup can be completed, along with evaluation of brainstem function to assess the associated Chiari II malformation. The long-term outcomes for these children have been reported in depth by McLone and coworkers (10). The survival rate for these children followed for 8 to 12 years was 85%, with 62% having an IQ of 80 or higher. With the development of clean intermittent bladder catheterization, 85% of children with myelomeningocele can achieve social continence of urine. The development of improved leg braces has allowed children with motor levels of at least L3 function to be community ambulators. McLone’s studies have shown that children with myelomeningocele can be functional and that the natural history of this disease is not one of relentless deterioration, as some prior studies had suggested. Any neurologic deterioration seen in children with myelomeningocele should be promptly investigated and treated.
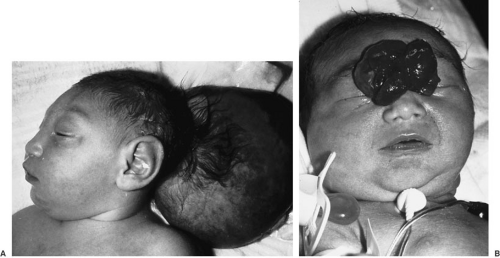 FIGURE 108-8. Encephalocele. (A) Infant with an occipital encephalocele. (B) Infant with a nasal encephalocele. |
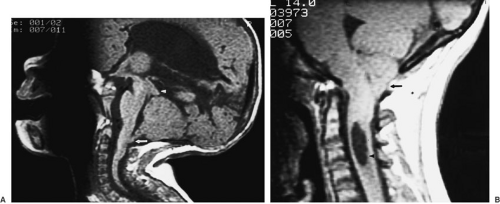 FIGURE 108-9. MR images of Chiari malformations. (A) Chiari II malformation. The cerebellar vermis is displaced caudad (arrow), and the midbrain tectum is beaked (arrowhead). (B) Chiari I malformation. In contrast to (A), the brainstem is normal, and only the cerebellar tonsils are descended into the cervical canal (arrow). A cervical syrinx (arrowhead) is present. |
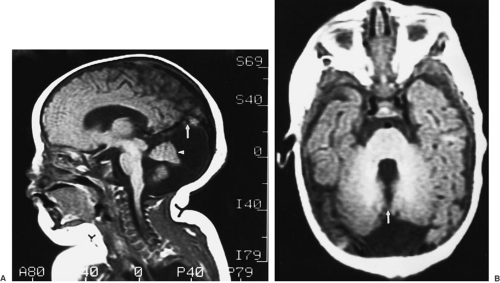 FIGURE 108-10. Magnetic resonance images of Dandy-Walker malformation. (A) Sagittal image shows an enlarged posterior fossa with a large cyst, elevated tentorium (arrow), and absent inferior cerebellar vermis. The superior vermis is indicated by an arrowhead. (B) Axial image shows absent inferior vermis and communication with the fourth ventricle (arrow). |
More recent advances in fetal diagnosis have made possible a current study of in utero closure of myelomeningocele at three U.S. centers. Early results suggest that in utero closure may result in a decreased need for CSF shunting, as well as reduction or even reversal of the Chiari II malformation (11). There is, however, significant risk to the fetus, including a 6% mortality due to extreme prematurity. The long-term benefits of in utero closure remain to be seen.
The congenital spinal cord lesions with intact skin are termed spina bifida occulta and share a common presentation, owing to tethering of the spinal cord. These lesions can also be associated with an overlying skin lesion, such as the dermal sinus, a subcutaneous lipoma, a hemangioma, or a hairy patch. The most common signs and symptoms of tethered cord include changes in gait, weakness, orthopedic deformity, and pain. The most common orthopedic deformities seen include varus and valgus, and cavus changes of the foot. In addition, recurrent hip dislocation and scoliosis may also be seen. Pain and sensory loss are common features of the tethered spinal cord, and both can be asymmetric and nondermatomal. In addition, changes in bowel and bladder function may be missed in young children. Neurologic dysfunction results from traction on the conus medullaris, with stretching and deformation of vessels overlying the tethered cord, resulting in ischemia of the conus (12). Spinal flexion or growth can result in chronic, repetitive ischemia to the spinal cord. With modern
magnetic resonance imaging (MRI) techniques, the diagnosis of spina bifida occulta is straightforward. The treatment of these lesions involves release of the tethered cord, with restoration of more normal anatomic relations of the spinal cord and surrounding structures. With early recognition and treatment, pain, sensory loss, and motor weakness are likely to improve, but if treatment is deferred until bowel or bladder dysfunction occurs, improvement of these functions is unlikely.
magnetic resonance imaging (MRI) techniques, the diagnosis of spina bifida occulta is straightforward. The treatment of these lesions involves release of the tethered cord, with restoration of more normal anatomic relations of the spinal cord and surrounding structures. With early recognition and treatment, pain, sensory loss, and motor weakness are likely to improve, but if treatment is deferred until bowel or bladder dysfunction occurs, improvement of these functions is unlikely.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


