Pediatric CNS tumors are classified according to the World Health Organization (WHO) classification.1,2 The assigned WHO grade describes the biology of the lesion, but a low grade does not always imply a good outcome. In this classification system, pediatric tumors are stratified according to the same criteria as those used for adults. As discussed in the following section, there are some limitations to this approach. Tumors classified as glioblastoma in children may, for example, be different biologically from tumors with similar morphology found in adults (Table 6.2). Cases that defy accurate classification despite best efforts may also be more common in children. Systemic metastases from brain tumors are highly unusual. Thus, in most cases, the main treatment strategy is focused on preventing or delaying local recurrence or to control growth. In some of the entities discussed in the following section, however, cerebrospinal fluid (CSF) dissemination is relatively common. Patients with ependymomas or medulloblastomas therefore will typically have imaging studies of the entire neuro-axis. Some patients including those with medulloblastoma will receive radiation treatment to the entire neuro-axis.

Pilocytic Astrocytoma
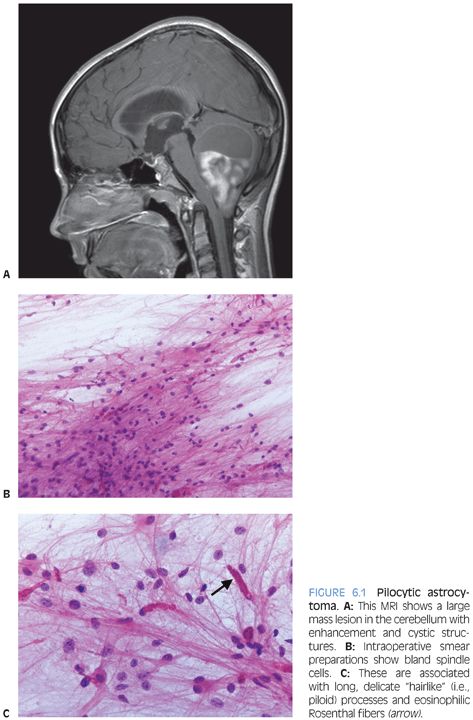
Pilocytic astrocytoma is a WHO grade I neoplasm that is most common in the first two decades of life. Common anatomic sites are the cerebellum, optic nerve/chiasm, and hypothalamus, but these tumors can be found virtually anywhere within the CNS. In some cases, like a patient with cerebellar pilocytic astrocytoma, surgery can be curative. In other patients, a hypothalamic tumor may slowly progress and ultimately be lethal. This example illustrates that we may consider certain tumors low-grade but that it can be very misleading to talk about “benign” brain tumors. Another uncommon but described phenomenon supporting this same point is the fact that patients with pilocytic astrocytoma may develop CSF dissemination.
Radiologically and grossly pilocytic astrocytomas are often associated with cyst formation. On enhanced magnetic resonance images, they typically exhibit enhancement (Fig 6.1). Prototypical cases are circumscribed with an expansile growth pattern. This can be a helpful diagnostic clue, but cases with more infiltrative edges are reported. Typical morphologic features (Fig. 6.2) of pilocytic astrocytoma include variation between dense and loose areas, presence of sometimes prominent hyalinized blood vessels, bipolar spindle cells with long processes, Rosenthal fibers, and sometimes eosinophilic granular bodies (EGBs). The presence of random atypical cells, degenerative changes with thrombosed vessels, organizing hemorrhage, necrosis, and mitotic figures may be worrisome or raise concern for other diagnoses. But these changes can all be part of the spectrum of pilocytic astrocytomas. Actual malignant progression in a pilocytic astrocytoma is described but highly unusual. Some cases may exhibit areas mimicking oligodendroglial differentiation.
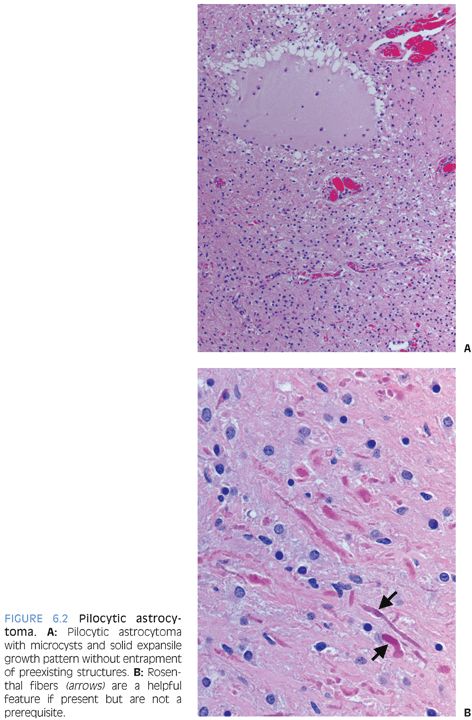
Depending on the morphologic features exhibited in a given case, the differential diagnosis may include the following: 1) Reactive piloid gliosis adjacent to either another tumor or another lesion such as a vascular malformation. 2) The glial component of a ganglioglioma may mimic pilocytic astrocytoma (see the following text). The identification of lesional dysmorphic ganglion cells allows the distinction. 3) Especially in small biopsy sample, the distinction from a low-grade diffuse astrocytoma may be challenging or impossible. Relative lack of clearly permeative invasive growth, presence of hyalinized blood vessels, Rosenthal fibers, EGBs, and knowledge of the radiologic appearance can all be helpful. Some small biopsies may, however, be best classified descriptively as “low-grade astrocytoma.” 4) In some cases, the unusual differential diagnosis may lie between a pilocytic astrocytoma with necrosis and prominent degenerative changes and a glioblastoma. Rare cases of “malignant” pilocytic astrocytoma with increased mitotic activity are described. 5) Pilocytic astrocytomas may have areas mimicking oligodendroglioma. Presence of areas with diagnostic morphologic features is usually key. Tumors with oligodendroglial differentiation are relatively rare in children and in infratentorial locations.
Special studies are of limited use in pilocytic astrocytomas. The lesional cells label for GFAP and S100 but these stains are rarely necessary. In some cases, staining for neurofilament may be helpful by demonstrating the lack of entrapped preexisting axonal processes. But this stain has to be interpreted with some caution because tumors are not always completely demarcated. Variants with more distinctly infiltrative growth are described. MIB-1 labeling is probably best avoided because the results may be more confusing than helpful. Some cases can go along increased labeling indices of 10% or more.4,5 Recent studies have shown BRAF rearrangements with tandem duplication and BRAF-KIAA1549 fusion in pilocytic astrocytomas.6,7 These are most common in the infratentorial tumors. The V600E mutation seen in melanomas is unusual in pilocytic astrocytomas but can be found in pleomorphic xanthoastrocytoma and ganglioglioma.8 In some unusual cases, fluorescence in situ hybridization (FISH) studies looking for these rearrangements may be helpful.
Pilomyxoid astrocytoma is closely related to pilocytic astrocytoma.9 It is most commonly found in the hypothalamus or chiasm of very young children. It is characterized by prominent myxoid matrix and angiocentric arrangement of lesional cells. Rosenthal fibers and EGBs are typically absent. These tumors tend to be more aggressive than pilocytic astrocytomas and are graded as WHO grade II.
Infiltrating Astrocytomas
Children, just like adults, develop tumors that are classified and graded in the WHO system as diffuse astrocytoma (WHO grade II), anaplastic astrocytoma (WHO grade III), and glioblastoma (WHO grade IV). The growth pattern of these lesions is characterized by individual cell infiltration between preexisting gray and white matter structures (Fig. 6.3). Because of this growth pattern, these tumors often show up grossly and radiologically as poorly demarcated areas of mass effect that may appear to be expanding preexisting structures. Enhancement is thought to often correlate with grade—it is usually absent in diffuse astrocytomas and associated with higher grade astrocytomas. It is reflective of the tumor containing blood vessels lacking normal blood–brain barrier.
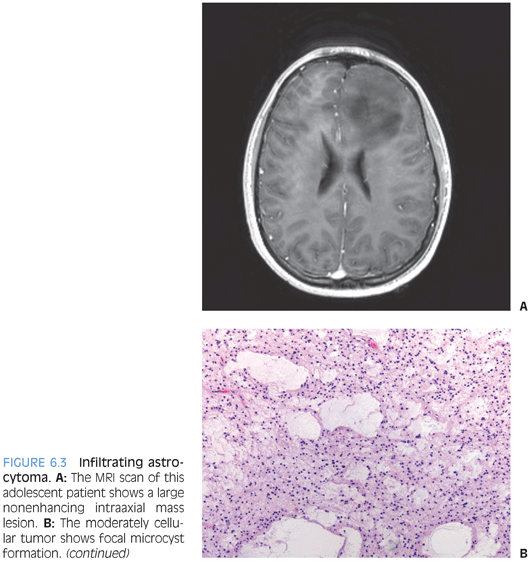

The diagnosis of these lesions often represents a two-step process. First, the tumor is classified as infiltrating astrocytoma and then the tumor is graded. The classification as infiltrating astrocytoma is based on the histologic growth pattern that goes along with the aforementioned entrapment of preexisting tissue elements. The background matrix typically has a fibrillary appearance representative of processes belonging to preexisting cells as well as tumor cells. The lesional cells morphologically exhibit features of astrocytic differentiation. In some cases, cells appear to consist of basically naked-appearing elongated and irregular-shaped nuclei. In other cases, cells may exhibit distinct eosinophilic and sometimes gemistocytic cytoplasm that often tapers out into processes.
The grading of these tumors occurs according to the same criteria as in adults. Increased proliferative activity with mitotic figures is required for a diagnosis of anaplastic astrocytoma. Endothelial proliferation or necrosis is required for classification as glioblastoma. The necrosis is often but not always pseudopalisading (Figs. 6.4 and 6.19). In some cases, MIB-1 labeling and p53 staining may provide some prognostic information.
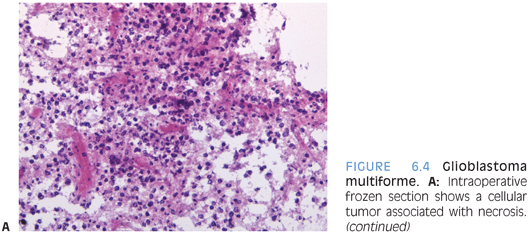
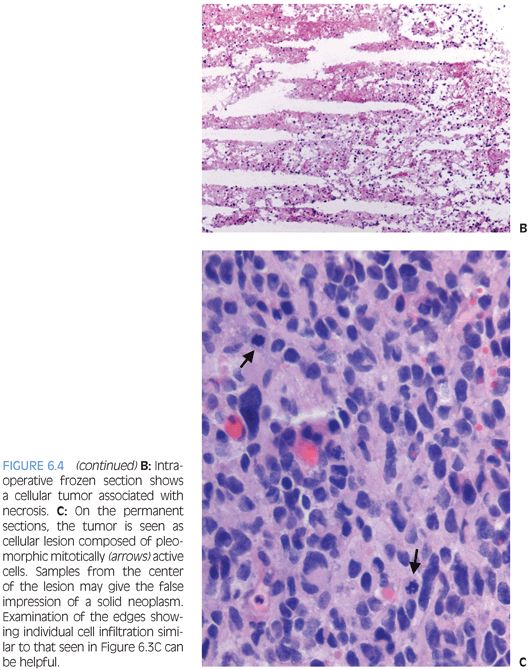
Special stains are of limited use in establishing the lineage of differentiation. Often, the tumor cells label for GFAP and S100. It is, however, important to remember that absence of GFAP expression does not exclude the diagnosis of glioblastoma. Neuronal markers such as neurofilament stain preexisting tissue elements. In some cases, negative staining for other markers can be helpful in excluding other entities that may be considered in the differential diagnosis, including lymphoma, systemic metastasis, or neuronal neoplasm.
The differential diagnosis varies depending on the grade of the tumor. Other high-grade tumors, such as primitive neuroectodermal tumors and atypical teratoid rhabdoid tumor, may be considered in the differential diagnosis of glioblastoma. In some cases, pleomorphic xanthoastrocytoma and pilocytic astrocytoma may mimic glioblastoma by showing pleomorphism, necrosis, or even mitotic activity. The final diagnosis in those cases rests on immunohistochemical and, in some cases, molecular studies. Reactive gliosis and other low-grade tumors including ganglioglioma may be considered in the differential diagnosis for low-grade lesions. The distinction of reactive gliosis may be difficult on biopsy samples. A history of disease processes that could illicit reactive gliosis or morphologic features of the same can be helpful. Uniform spacing of glial cells, lack of frank atypia, reactive vascular changes, and macrophage infiltration can be suggestive of reactive etiology.
When grading and classifying astrocytomas, we often treat pediatric patients like little adults. Molecular studies suggest that this approach has limitations. Glioblastomas in children are associated with different molecular changes than those typically seen in their adult counterparts.10 There may even be differences between glioblastomas of early childhood and older children. In adult patients, studies looking for isocitrate dehydrogenase (IDH)1/IDH2 mutations and epidermal growth factor receptor (EGFR) amplification are sometimes employed. IDH1/IDH2 mutations and EGFR amplification are rare in pediatric astrocytomas.11 Platelet-derived growth factor receptor, α-polypeptide (PDGFRA) amplification is relatively common in pediatric glioblastomas but rare in adult cases. Recent data suggests that about a third of pediatric glioblastomas show mutations in the H3F3A gene encoding the replication-independent histone 3 variant H3.3.11,12 This leads to a unique methylation signature in the cancer genome.11
Oligodendroglioma
In adults, oligodendrogliomas are a well-defined group of tumors that exhibit an infiltrating growth pattern similar to that seen in infiltrating astrocytomas but distinctly different cytomorphologic features. The lesional cells show round regular nuclei. On paraffin sections, they often show perinuclear halos as a processing artifact lacking on frozen sections. Sometimes, cells with round regular nuclei but distinct eosinophilic cytoplasm are seen, so-called mini-gemistocytes. Oligodendrogliomas show a strong association with codeletion of 1p and 19q—some would argue they are defined by these molecular changes.
Sometimes, prototypical oligodendrogliomas with 1p/19q codeletion are seen in older children. In younger patients, these tumors are uncommon. Rare tumors in these patients with oligodendroglioma morphology typically lack the 1p/19q codeletion.
Pleomorphic Xanthoastrocytoma
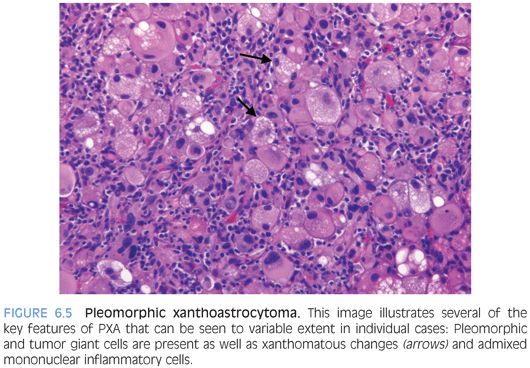
These are low-grade tumors that present as enhancing superficial lesions. Clinically, they are often associated with seizures.13,14 Most patients are in the second or third decade of life. In some cases, surgical resection can be curative. Pleomorphic xanthoastrocytomas (PXAs) are most often seen in the hemispheres as superficial lesions that may exhibit striking extension into the subarachnoid space resulting in a meningocerebral distribution. These tumors are appropriately named for some of their key morphologic features (Fig. 6.5). 1) They are composed of cells exhibiting features of astrocytic differentiation. 2) Often, they show marked pleomorphism. 3) Some cells may show distinct vacuolated foamy xanthomatous cytoplasm attributable to cytoplasmic lipid. EGBs are found in virtually all cases. Other features are an overall expansile growth pattern and at least focal distinct pericellular reticulin. PXAs are typically classified as WHO grade II despite the pleomorphism and often high cellularity that may at first glance be worrisome features. Higher grade variants are very unusual but described. Rare ganglion cells may be found, and in some cases, the distinction from ganglioglioma may be difficult.
Subependymal Giant Cell Astrocytoma
Subependymal giant cell astrocytomas (SEGAs) arise in the wall of the lateral ventricles and are virtually always tuberous sclerosis–associated discrete tumors.15 They are most common in the second decade of life. Because of their location, these tumors may cause obstruction of CSF flow at the level of the foramen of Monroe. Radiologically, these are solitary or bilateral demarcated enhancing tumors. The lesional cells vary in appearance from spindled to plumb and from small to large (Fig. 6.6). Calcifications are common. Necrosis, increased cellularity, and nuclear atypia can be seen in this WHO grade I tumor. The lesional cells may express GFAP or neuronal markers such as synaptophysin. Sometimes, markers of both lineages may be expressed in an individual cell. Because of the mixed differentiation, these tumors are sometimes referred to as subependymal giant cell tumor. The histologic features are indistinguishable from those found in the subependymal nodules of tuberous sclerosis that may form multinodular changes in the ventricular wall likened to candle drippings. Size is the distinguishing feature. The underlying molecular alteration driving the growth of these tumors is activation of the mammalian target of rapamycin (mTOR) signaling pathway. Therefore, patients are often treated with rapamycin to control tumor growth.

Astroblastoma
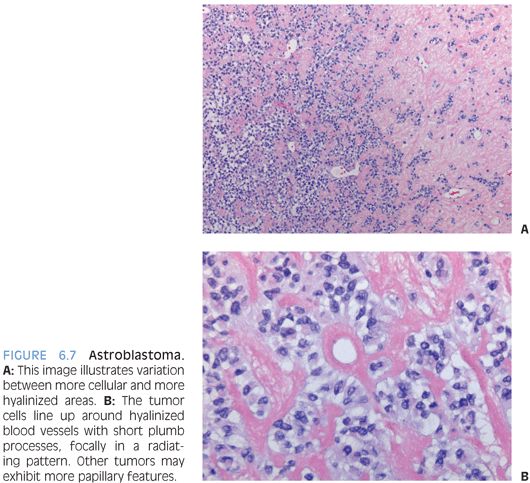
This is a well-demarcated solid or cystic tumor that presents as contrast-enhancing superficial hemispheric lesion.16 Despite the name, these tumors are not immature blastic but exhibit some ependymal features. The lesional cells show perivascular arrangement that may mimic ependymal pseudorosettes but typically goes along with shorter, more plumb stubby cell processes (Fig. 6.7). Often, there is distinct vascular/perivascular hyalinization. GFAP and S100 are strongly positive, and epithelial membrane antigen (EMA) staining may also be seen. Intercellular junction and microvillous processes as found in ependymomas may be present. Often, these tumors behave as low-grade lesions, but more aggressive cases are reported, and a definitive grade has not been assigned in the WHO system. Ependymoma and even papillary meningioma may be considered in the differential diagnosis.
Desmoplastic Infantile Ganglioglioma/Astrocytoma
Desmoplastic infantile ganglioglioma/astrocytomas (DIG/DIAs) is a WHO grade I tumor that usually presents as large hemispheric lesion in early childhood.17,18 The tumor is contrast enhancing on magnetic resonance imaging (MRI) and is often associated with cystic changes. A key morphologic feature is the desmoplasia that appropriately has become part of the entity’s name. Prominent collagen bundles are seen admixed with the tumor and may in places crowd out tumor cells. They are highlighted on a trichrome stain. Nestled between the desmoplastic collagenous areas are nests, strands, or islands with fine fibrillary background (Fig. 6.8). The lesional cells are small astrocytes with bland plump oval nuclei. The presence of a neuronal component distinguishes DIG from DIA. Sometimes, the neuronal component can at least in part take the form of larger ganglion cells, but often, the neuronal cells are small and therefore difficult to distinguish from lesional astrocytes on the hematoxylin and eosin (H&E) stain. Staining for neuronal markers can therefore be helpful. The glial component is positive for GFAP. In rare cases, a cellular mitotically active small cell component can be present. This may mimic primitive neuroectodermal tumor–like differentiation. This latter feature does, however, not clearly indicate poor outcome and is at the moment of undetermined significance.

Dysembryoplastic Neuroepithelial Tumor
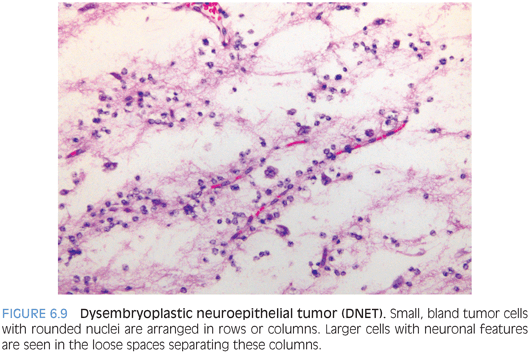
These are WHO grade I glioneuronal lesions that typically arise superficially in the hemispheres early in life.19,20 Seizures are a common presenting feature. Grossly and radiologically, the tumor often appears as multinodular, superficial, and, at least partly, intracortical lesion. The histologic appearance is that of a tumor that may mimic oligodendroglioma because the dominant cell population is composed of small cells with round nuclei often surrounded by perinuclear halos on paraffin sections. These cells tend to be arranged in rows around vessels and bundles of processes leaving small paucicellular spaces filled with mucinous material. Larger ganglion cells floating in these mucinous pools are termed “floating neurons” and represent a helpful feature (Fig. 6.9). Sometimes, more complex patterns with areas mimicking pilocytic astrocytomas or diffuse astrocytomas are described. The low-power multinodular appearance, the arrangement of the small oligodendroglioma-like cells in rows or columns, and the presence of floating neurons are key features for the diagnosis. The absence of 1p/19q codeletions can be helpful at times to exclude the possibility of oligodendroglioma.
Ganglioglioma
Ganglioglioma is typically classified as WHO grade I tumor. These are most frequently seen as hemispheric tumors, often in the temporal lobes,21 but they can be found virtually anywhere in the CNS. Seizures are a common presenting feature. Grossly and radiologically, they may be solid or cystic. Gangliogliomas are one of a set of low-grade tumors that can present with an MRI showing a cystic lesion with an enhancing mural nodule (e.g., as also seen in pilocytic astrocytomas or hemangioblastomas). Typically, these are tumors with solid expansile growth pattern. The neuronal component of this glioneuronal tumor consists of large, often dysmorphic, ganglion cells (Fig. 6.10). The spacing of the neurons is haphazard, and abnormally clustered “kissing” neurons may be seen. The glial component can be more variable and may mimic pilocytic astrocytoma, diffuse astrocytoma, or even oligodendroglioma. EGBs, calcifications, and perivascular lymphocytes are common features and helpful clues. Immunohistochemical studies for neuronal markers can confirm the differentiation of the neuronal component. In some cases, the distinction between lesional neurons and neurons entrapped by an infiltrating glioma may be difficult. Dysmorphic ganglion cells in cortical dysplasia may mimic those of ganglioglioma but more closely follow normal anatomic distribution.




