another family member who has survived with a cardiac anomaly, more often, there is no family history. Inheritable or even acquired defects in a single gene (e.g., Marfan syndrome) or a chromosomal aneuploidy (e.g., trisomy) is not yet identifiable in most patients with cardiac malformations. More commonly, there may be susceptibility from inherited or acquired mutations in two or more genes, perhaps with additional alterations in gene transcription or posttranscriptional processes from maternal-fetal folate metabolism or fetal exposure to specific pharmacologic, biochemical, infectious, and environmental factors that cumulatively surpass a threshold of liability (9,10,11,12). These result in pathogenic changes in embryonic development, including the following:
TABLE 30.1 Neonatal (First Month) Cardiac Diagnostic Distributiona | ||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||
Defects in mesenchymal tissue migration (tetralogy of Fallot, truncus arteriosus, interrupted aortic arch, malalignment conal-septal ventricular septal defects, transposition of great arteries)
Extracellular matrix defects (endocardial cushion defects)
Abnormal cell death (muscular ventricular septal defect, Ebstein anomaly)
Targeted growth (anomalous pulmonary vein connection, single atrium)
Defective situs and cardiac looping (heterotaxy syndromes, l-transposition)
TABLE 30.2 Infant (First-Year) Cardiac Diagnostic Distribution and Mortalitya | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Down syndrome and endocardial cushion defects, Williams syndrome and supravalvar aortic stenosis, DiGeorge syndrome and tetralogy of Fallot, truncus arteriosus, and interrupted aortic arch (Table 30.3). Recognition that a child has a syndrome associated with a cardiac anomaly or vice versa should prompt an investigation for possible associated anomalies (16,17,18,19,20).
TABLE 30.3 Congenital Disorders Associated with Cardiac Disease | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 30.4 Incidence of Severe Associated Noncardiac Anomalies Among 2,220 Infants with Heart Disease | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
TABLE 30.5 Possible Teratogens for Congenital Heart Disease | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
 FIGURE 30.1 Fetal circulation is in parallel, and the amount of blood handled by the left and right ventricles is 125 and 90 mL, respectively. Only 40 mL passes through the aortic arch to the descending aorta, and only a small fraction passes through the lungs. The numbers inside the diagram represent relative blood flow (mL); the numbers in italics are pressure measurements. Modified from McElhinney DB, Tworetzky W, Lock JE. Current status of fetal cardiac intervention. Circulation 2010;121:1256. |
circulation. Oxygen saturation in the ascending aorta rises from about 65% in the fetus, to about 93% immediately after birth. The ductus venosus functionally closes, establishing the portal circulation as an independent loop between two capillary beds. With removal of the low-resistance placenta, systemic vascular resistance increases. The relative fall in the pulmonary and rise in the systemic vascular resistance result in a transitory left-toright shunt through the ductus arteriosus. In 50% of term babies, the ductus is completely constricted by the end of the first day of life and normally becomes anatomically obliterated at about 10 days of age. Even among cyanotic newborns who are duct dependent, the ductus may inexorably close, often severing the infant’s only source of pulmonary or systemic blood flow. The mechanisms causing closure of the ductus arteriosus are not completely understood but involve decreased prostaglandins and increased blood oxygen. Prostaglandin levels in the blood decrease at birth as a result of removal of their placental source of production and increase in perfusion of the lungs, where prostaglandins are metabolized.
 FIGURE 30.2 Mature circulation is in series, and the amount of blood carried by the two ventricles is approximately the same as before birth. The lungs carry an amount equivalent to the cardiac output, as does the ascending aorta. The numbers inside the diagram represent relative blood flow (mL); the numbers in italics are pressure measurements. |
TABLE 30.6 Indications for Fetal Echocardiography | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
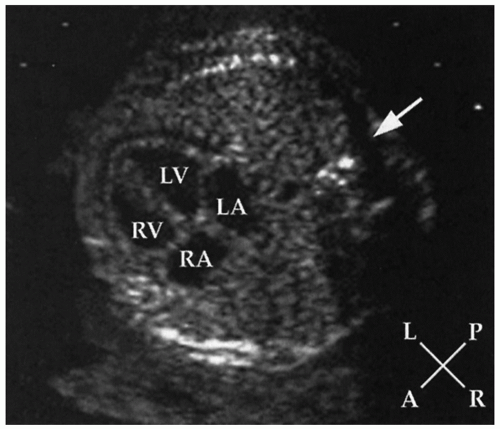 FIGURE 30.3 Echocardiogram of the normal fetal heart in a four-chamber view demonstrating the position of the heart and the cardiac chambers in a cross-section of the chest. A, anterior; L, left; LA, left atrium; LV, left ventricle; P, posterior; R, right; RA, right atrium; RV, rightventricle; arrowdenotes the spine. |
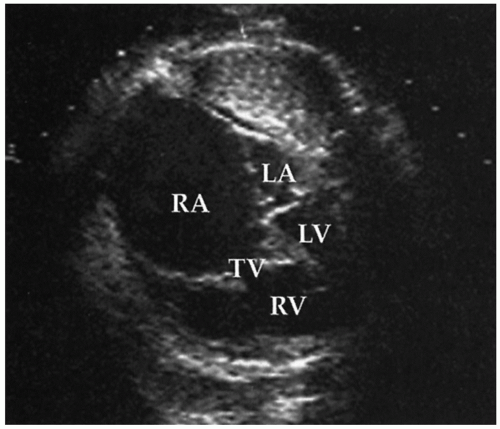 FIGURE 30.4 Echocardiographic cross-sectional views of the fetal chest in an infant with Ebstein anomaly of the tricuspid valve. The right atrium (RA) is markedly dilated and fills much of the thorax. The left atrium (LA) and left ventricle (LV) are of normal size but are dwarfed by the right-sided structures. The severely regurgitant tricuspid valve has apical displacement of the septal leaflet into the right ventricle (RV). |
small fetal cardiac size, rapid heart rate, and lack of ECG gating (38,39). Severe ventricular dysfunction may manifest as generalized hydrops fetalis, which is readily recognized by ultrasound as pleural and peritoneal fluid accumulation and cutaneous edema.
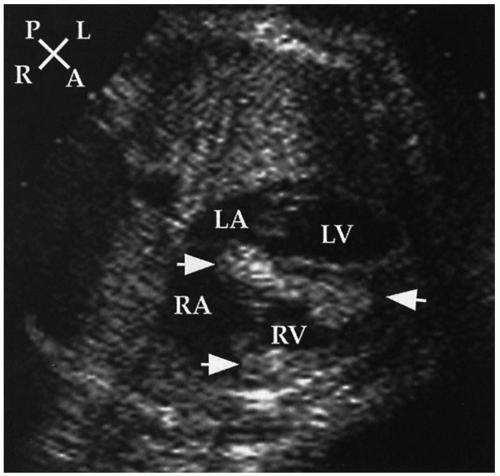 FIGURE 30.5 Echocardiographic cross-sectional view of a fetus with multiple intramyocardial rhabdomyomas ( arrows). The infant was subsequently diagnosed with tuberous sclerosis. A, anterior; L, left; LA, left atrium; LV, left ventricle; P, posterior; R, right; RA, right atrium; RV, right ventricle. |
 FIGURE 30.6 Echocardiogram of a fetus with a premature atrial beat. The upper left image is a two-dimensional view with a cursor (line) through the fetal right atrium (RA) and aortic valve (AOV), demonstrating the axis of the simultaneous M-mode echocardiogram seen in the lower panel. The M-mode tracing depicts the motion of the fetal right atrial wall and aortic valve in the cursor line over a time frame of 3 seconds. A series of normal atrial wall contractions are interrupted by a premature contraction (large arrow) followed by opening of the aortic valve (small arrow), demonstrating a premature atrial beat conducted to the ventricle. LV, left ventricle; RV, right ventricle. |
the initial evaluation. Any one of these alone is rarely diagnostic. A number of lesions result in cyanosis; quite a number of lesions are also associated with loud murmurs; others are associated with little or no murmur; some cause shock (see Figs. 30.7 and 30.8). Others have chest radiographs with increased pulmonary arterial or venous markings; others have diminished pulmonary vascular markings. Most have an undistinguished ECG at birth; while some have left-axis deviation on ECG (see Figs. 30.9 and 30.10). Most cardiac anomalies vary in their characteristics at presentation. Furthermore, often it is not possible to determine with certainty if the second heart sound is split or not, or if the pulmonary vascular markings on chest radiograph are normal versus increased or normal versus decreased. Clinical analysis requires weighting of the categories of evidence as to its certainty and other possibilities. A classical diagnostic approach based on sequential analysis of data categories is limited by these types of weaknesses in the clinical information and is no stronger than the weakest link in the chain of information. However, interweaving of the findings provides a matrix of diagnostic information that remains intact even when one category of findings is weak. Overlapping possible anomalies suggested from history, physical examination, chest radiograph, and ECG, as if with a series of Venn diagrams (see Figs. 30.7, 30.8, 30.9, and 30.10), provides information that allows a careful observer to quickly determine which anomaly, or which two or three possible anomalies, may be present. Comparing the anomalies consistent with the clinical presentation with the anomalies consistent with the murmur findings, other physical exam findings, chest radiograph findings, and electrocardiographic findings usually focuses the list of possible anomalies on one or two primary choices (see Fig. 30.11). This may provide an important advantage in the timely and efficient management of potentially lifethreatening anomalies. For example, the combination of cyanosis, soft or no murmur, single S2, chest radiograph with decreased pulmonary vascular markings and normal heart size, and ECG R axis of 50 degrees suggests pulmonary atresia, which is an anomaly in which life depends upon maintaining ductal patency (see Figs. 30.8 and 30.10). Two-dimensional echocardiogram should be obtained promptly if significant cardiac disease is suspected. This technique when done by personnel trained for evaluation of congenital cardiac anomalies in neonates accurately demonstrates the
anatomy, occasionally uncovering a potentially lethal lesion before symptoms. Appropriate initial management (e.g., infusion of PGE1 in a cyanotic infant suspected to have pulmonary atresia) need not await availability of echocardiography (see Fig. 30.12 and Management Procedures for Severe Cardiac Disease).
 FIGURE 30.7 The differential diagnosis of cardiac exam findings in acyanotic neonates. Anomalies in larger print are more common. +/-, sometimes; up arrow, increased; down arrow, decreased; AP window, aorticopulmonary window; AS, aortic stenosis; ASD, atrial septal defect; CAVC, complete atrioventricular canal defect; CoAo, coarctation of the aorta; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomopathy; Interrup. Ao, interrupted aortic arch; MS, mitral stenosis; PDA, patent ductus arteriosus; PS, pulmonary stenosis; VSD, ventricular septal defect. |
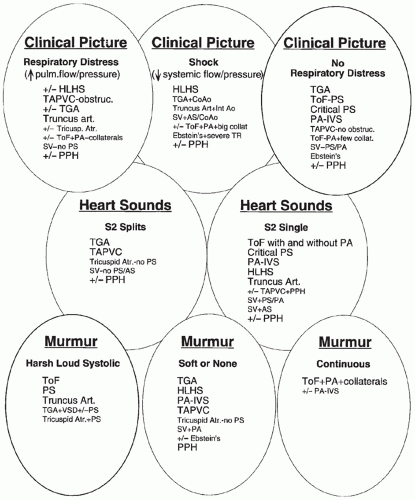 FIGURE 30.8 The differential diagnosis of cardiac exam findings in cyanotic neonates. +/-, sometimes; up arrow, increased; down arrow, decreased; AS, aortic stenosis; CoAo, coarctation of aorta; collat., systemic to pulmonary artery collateral vessels; Ebstein’s, Ebstein anomaly of the tricuspid valve; HLHS, hypoplastic left heart syndrome; Int Ao, interrupted aorta; PA, pulmonary atresia; PA-IVS, pulmonary atresia with intact interventricular septum; PPH, persistent pulmonary hypertension syndrome; PS, pulmonary stenosis; SV, single ventricle; TAPVC, totally anomalous pulmonary venous connection; TGA, transposition of great arteries; ToF, tetralogy of Fallot; tricuspid atr., tricuspid atresia. |
 FIGURE 30.9 The differential diagnosis of chest radiographic and electrocardiographic findings in acyanotic neonates. Abbreviations, see Figure 30.7; CXR, chest x-ray; LAD; left axis deviation; LV, left ventricle; Pulm. vasc. markings, pulmonary vascular markings; RV, right ventricle. |
various anomalies but is often not proportional to the severity of the lesion. The absence of a murmur does not preclude serious heart disease. To the contrary, many life-threatening cardiac anomalies may be associated with little or no murmur. In a neonate with cyanosis and/or shock and suspected cardiac anomaly, the presence of little or no murmur provides a diagnostic clue (see Figs. 30.7 and 30.8). The pitch of a murmur is associated with the pressure gradient across the abnormality causing the murmur. Tiny ventricular septal defects develop a characteristic fairly high-pitched murmur when the right ventricular pressure decreases to much less than the left ventricular pressure. Severe pulmonary or aortic stenosis can sometimes be distinguished from mild stenosis by a high-pitched harsh loud murmur and an associated thrill.
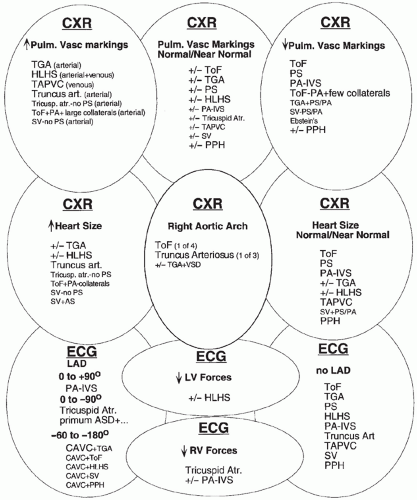 FIGURE 30.10 The differential diagnosis of chest radiographic and electrocardiographic findings in cyanotic neonates. Abbreviations, see Figs. 30.8 and 30.9. |
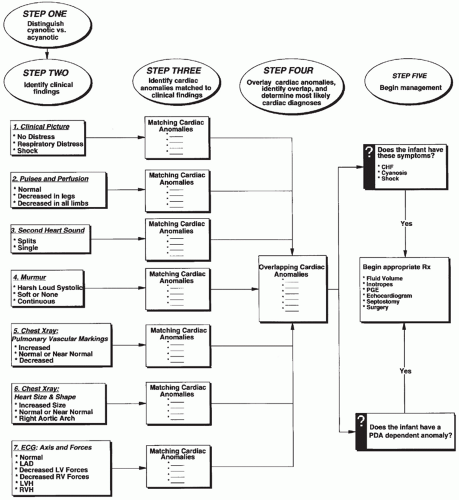 FIGURE 30.11 A process for diagnosing cardiac anomalies from findings on cardiac exam, chest radiograph, and electrocardiogram. ASAP, as soon as possible; c/w, consistent with; FiO2, fractional percentage of inspired oxygen; PDA, patent ductus arteriosus. |
Poor mixing of separate parallel circulations (e.g., transposition of the great arteries
Other anomalies with transposition physiology such as Taussig-Bing-type double-outlet right ventricle
Restricted pulmonary blood flow and right-to-left shunting of unoxygenated systemic venous blood to the systemic arterial circulation (e.g., tetralogy of Fallot, critical pulmonary stenosis, tricuspid atresia)
Right-to-left shunting from intracardiac mixing with normal or increased pulmonary blood flow (e.g., total anomalous pulmonary venous connection without obstruction, truncus arteriosus, single ventricle without pulmonary stenosis, hypoplastic left heart syndrome)
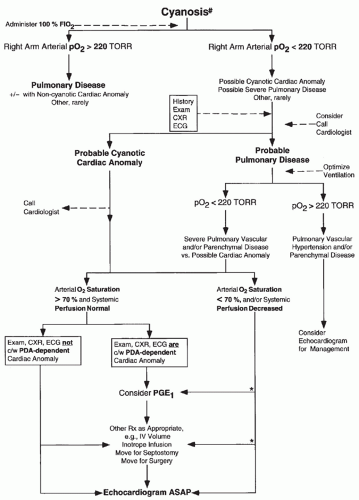 FIGURE 30.12 An approach to the diagnosis and management of cyanotic infants. #, cyanosis; see text for additional details of assessment of cyanosis; +/-, possibly; *, see text concerning management of specific anomalies; ASAP, as soon as possible; c/w, consistent with; FiO2, fraction inspired oxygen; Rx, treatment. |
TABLE 30.7 Top Five Diagnoses Presenting at Different Ages | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
cardiothoracic ratio greater than 0.6 in an anterior-posterior projection in the presence of an adequate inspiration. The aortic arch position can be assessed, even in the presence of a large overlying thymus, by deviation of the trachea to the opposite side. Associated noncardiac anomalies that provide clues to the cardiac diagnosis may be discovered by radiographic findings, for example, heterotaxy (asplenia syndrome, malrotation), absence of the thymus gland (DiGeorge syndrome), vertebral anomalies (VACTERL association), and abnormal sternal ossification (Down syndrome).
TABLE 30.8 Normal Maturational ECG Changes | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
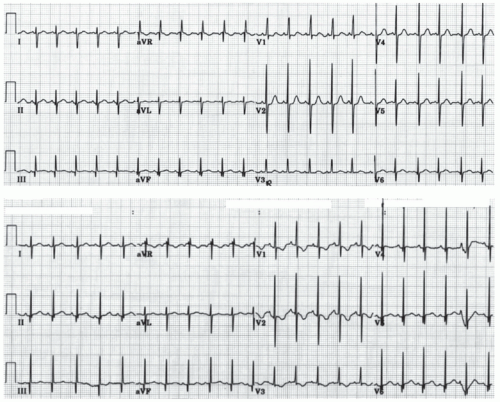 FIGURE 30.13 Electrocardiograms from healthy ½ day old (top) and 5 day old infants (lower) demonstrate normal neonatal repolarization changes. V1 T wave morphology changes from upright to inverted. |
Within the first days of life, there are significant changes in repolarization, including T axis and rate-corrected QT interval (QTc), as well as changes in R and S wave amplitudes, that influence interpretation. Compared with a 1 year old, 1 day old babies normally have relatively fast heart rates (93 to 154, average 123/minute), relatively rightward R axis (60 to 195, average 123 degrees) and relative right ventricular hypertrophy (R in V1 5 to 16 mm) (44). During the first 4 days of life a variably longer QTc, an evolution of changes in T wave polarity and voltage (e.g., in V1 from upright to inverted), and nonspecific ST segment changes are common. After age 4 days, QTc is 440 milliseconds or less in 97.5% of newborns but can be much longer with electrolyte abnormality (e.g., hypocalcemia, hypokalemia), drug effect, brain injury and genetic prolonged QT syndromes.
intravenous or umbilical artery catheters can sometimes serve as a useful adjunct to color Doppler in detection of shunts.
 FIGURE 30.14 Echocardiographic parasagittal parasternal view of a patent ductus arteriosus. A: Doppler analysis demonstrates flow away from the transducer within the pulmonary artery and aortic isthmus and, in white, a jet of flow toward the transducer through the patent ductus arteriosus into the pulmonary artery. B: Quantification of the velocity of the flow jet through the ductus arteriosus with a continuous-wave Doppler technique and application of the Bernoulli principle allows the aortic-to-pulmonary-artery systolic pressure gradient to be measured. The pressure gradient by this technique is 4 × (maximum instantaneous velocity)2. The pulmonary artery peak systolic pressure can be estimated by the difference in the arterial systolic pressure and the pressure gradient across the ductus arteriosus. AO, aorta; DAO, descending aorta; LA, left atrium; MPA, main pulmonary artery; PDA, patent ductus arteriosus. |
 FIGURE 30.15 A: Echocardiographic apical four-chamber view in systole. Color Doppler analysis of the right heart demonstrates a jet of tricuspid regurgitation depicted by the blue flow jet (white arrow). B: Quantification of the velocity of the regurgitant jet by application of continuous-wave Doppler technique along the axis of the dotted line in the upper panel. Application of the measured triscuspid regurgitant velocity within the Bernoulli equation allows the right-ventricle-to-rightatrial peak pressure gradient to be measured and estimation of the right ventricular pressure. LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle; TR tricuspid regurgitation. |
of Fallot and pulmonary atresia? Is surgical unifocalization or catheter closure of the specific collaterals preferable? What is the anatomy of the coronary arteries in the patient with pulmonary atresia and intact ventricular septum? What is the anatomy of the coronary arteries with tetralogy of Fallot or transposition where abnormality is suspected and/or echocardiographic imaging is nondiagnostic? In selected patients with cardiomyopathy, light and electron microscopic analysis of ultrastructural anatomy and biochemical analysis of myocardium obtained by biopsy may uniquely provide a diagnosis. In many situations, diagnostic catheterization may be safer or more useful after initial palliative surgery, such as in hypoplastic left heart syndrome or following shunt procedures in those with complex intracardiac anomalies and pulmonary atresia.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


