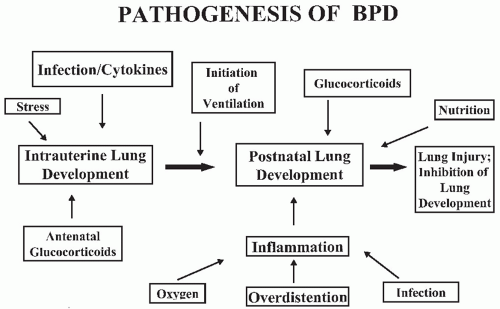
No single factor has been identified as the cause of BPD. Its origin is multifactorial and may depend on the nature of the injury, mechanisms of response, or the infant’s inability to respond appropriately to the injury process and “repair” the lung (
Fig. 27.1). While Northway attributed the occurrence of BPD primarily to prolonged hyperoxia in infants with RDS, numerous other causes have been proposed.
Barotrauma/Volutrauma
Although the initial phases of lung injury in BPD are the result of the primary disease process (e.g., RDS), superimposed positive-pressure mechanical ventilation appears to add to the lung injury and provoke a complex inflammatory cascade that ultimately leads to chronic lung disease. Barotrauma is the term generally used to describe the lung injury that occurs secondary to positive-pressure mechanical ventilation, although volutrauma from excessive tidal volume ventilation may be a more appropriate term to describe this lung injury process. The role of volutrauma in BPD depends on several factors, including the structure of the tracheobronchial tree and the physiologic effects of surfactant deficiency. When surfactant deficiency is coupled with an immature epithelium, surface tension forces are elevated, aeration is unequal, and most terminal alveoli are largely collapsed. The pressure needed to distend these poorly compliant saccules is high and is transmitted to the terminal bronchioles and alveolar ducts. In the premature infant, these airways are highly compliant and subject to rupture. Gas then dissects into the interstitium and pleural space, resulting in the development of pulmonary interstitial emphysema (PIE) and pneumothorax. These complications are strongly associated with the development of BPD, suggesting that ventilatorinduced lung injury is important in the pathogenesis (
12). Brew et al. (
13) have shown that even brief periods of positive-pressure ventilation cause significant damage to the immature lung, with the injury focused on the terminal bronchioles. However, in the absence of continued ventilation, the lung is able to repair itself, suggesting that less invasive forms of respiratory support may be beneficial in the preterm newborn. Even with normal tidal volumes, ventilation of the immature or injured newborn lung results in nonuniform inflation and relative overdistension of ventilated segments, especially in the presence of low functional residual capacity (FRC) due to surfactant deficiency. Excessive stretching of capillary endothelium and distal lung epithelium is associated with the release of reactive oxygen species (ROS), marked inflammatory changes in the lung, damage to cellular tight junctions, and increased permeability to serum proteins that may further inhibit surfactant function, creating a vicious cycle that promotes cell damage and ultimately lung injury (
14,
15,
16,
17). Although the adverse effects of tidal volume breaths in lungs with low FRC can be ameliorated with better lung recruitment by application of higher positive end-expiratory pressure (PEEP), experimental studies have shown that overdistension (not increased pressure) is responsible for lung injury in the surfactant-deficient lung. Strapping the chest wall of a ventilated animal to restrict overexpansion permits the generation of increased lung pressure while limiting excessive lung stretch and volume. These studies found markedly reduced lung injury with lower tidal volumes, reinforcing the concept that excessive volume and not pressure is the primary cause of lung injury in animal models (
18).
Strategies to prevent lung injury have focused on the use of exogenous surfactant as well as various modes of respiratory support. Golembek and Truog have presented a comprehensive review of optimal ventilatory strategies in
Chapter 28, which should be consulted. However, while the widespread use of surfactant has reduced some complications of mechanical ventilation (by reducing airway pressures and air leak while improving lung recruitment), BPD continues to be an important problem, suggesting that barotrauma/volutrauma is only one of many factors involved in the pathogenesis of BPD.
Oxygen and Antioxidants
Under normal conditions, a delicate balance exists between the production of ROS and the antioxidant defenses that protect cells
in vivo. ROS are molecules with extra electrons in their outer ring, and they are toxic to living tissues (
Table 27.2). Oxygen
has a unique molecular structure and is abundant within cells. It readily accepts free electrons generated by oxidative metabolism within the cell, producing ROS. Increased ROS production can occur under conditions of hyperoxia, reperfusion, or inflammation. Alternatively, ROS can increase because of an inability to quench production because of inadequate antioxidant defenses. Damage caused by ROS includes lipid peroxidation, mitochondrial injury, protein nitration, and unraveling of nucleic acids.
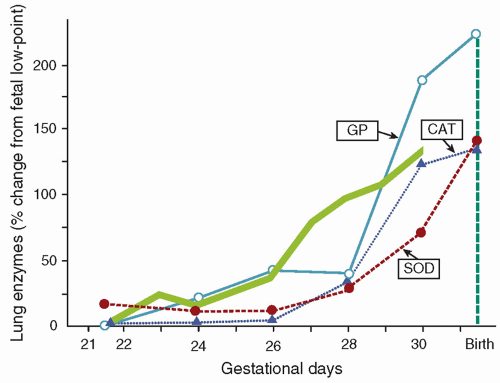
The premature neonate may be more susceptible to ROS-induced injury because adequate concentrations of antioxidants may be absent at birth. Frank and associates documented the development of the antioxidant enzymes superoxide dismutase (SOD), catalase, and glutathione peroxidase in the lungs of rabbits during late gestation (
Fig. 27.2) (
19). The 150% increase in these enzymes during the last 15% of gestation parallels the maturation pattern of pulmonary surfactant. These developmental changes in the fetal lung allow proper ventilation by reducing surface tension and provide for the transition from the relative hypoxia of intrauterine development to the oxygen-rich extrauterine environment. Premature birth can occur before the normal up-regulation of these antioxidant systems and transfer of other nonenzymatic antioxidants (e.g., vitamin E, ascorbic acid, glutathione, and ceruloplasmin) and may result in an imbalance between oxidants and antioxidants and an increased risk for developing BPD (
20).
Clinical studies suggest that ROS are involved in the pathogenesis of BPD. Plasma concentrations of allantoin (oxidative by-product of uric acid), expired pentane and ethane (ROS-induced lipid peroxidation), protein carbonyl formation (oxidation of proteins), and plasma 3-nitrotyrosine (reactive nitrogen species; endogenous nitric oxide reacting with superoxide) have all been shown to be significantly elevated in the 1st week of life in infants developing BPD compared to infants who recover without significant lung injury (
21,
22,
23,
24). The multicenter supplemental therapeutic oxygen for prethreshold-retinopathy of prematurity (STOP-ROP) trial examined whether exposing premature infants to higher inspired oxygen concentrations would prevent the development of severe ROP. While the effects of the increased oxygen were minimal on the eyes, exposed infants had a 55% increase in the incidence of BPD and pulmonary infections (
25). Even brief periods of hyperoxia may be associated with an increased risk of significant lung injury. Vento et al. (
26) randomized preterm infants needing resuscitation to 30% or 90% oxygen. Infants initially receiving 90% oxygen (for only a few minutes) required more respiratory support, had a higher incidence of BPD, and had more evidence of oxidation and inflammation than infants receiving 30% oxygen. Finally, Saugstad (
27) conducted a meta-analysis of multiple clinical studies of preterm infants and found a strong association between oxygen exposure and the development of BPD.
Further evidence for the role of ROS in lung injury comes from animal studies demonstrating that exposure to chronic hyperoxia can induce oxidant injury, subsequent inflammation and lung injury that has many features seen in infants developing BPD. Both epithelial and endothelial cells are extremely susceptible to oxidant injury, leading to increased permeability edema and cell dysfunction. Hyperoxia also impairs mucociliary function (increasing the susceptibility to infection), promotes inflammation, and inactivates antiproteases, further complicating clinical outcome. Other animal studies have found that antioxidant supplementation with SOD and catalase reduces cell damage, increases survival, and prevents lung injury from prolonged hyperoxia and mechanical ventilation (
28,
29). Genetically engineered mice overexpressing SOD survive longer, whereas mice with disrupted SOD genes die more quickly in a hyperoxic environment compared to normal diploid controls (
30,
31). These studies all demonstrate that ROS are intimately involved in the development of acute and chronic lung disease in newborn infants.
Inflammation
Marked inflammation in the lung appears to play an important role in the pathogenesis of BPD and allows many factors to be unified into a single common pathway of this disease. The initial stimuli activating the inflammatory process in the lung may be ROS, excessive stretch, infectious agents, or other stimuli that result in the attraction and activation of leukocytes. Inflammatory mediators and cellular responses have been found to be prominent in animal models of lung injury and in infants who develop BPD, possibly through activation of the critically important transcription factor, nuclear factor-κB (
32).
A variety of biomarkers associated with the inflammatory process have been identified in tracheal aspirates, serum, and urine which have been consistently linked to the development of BPD (
32). Bose et al. (
33) analyzed serum samples from 932 extremely preterm infants enrolled in the extremely low gestation age newborn (ELGAN) study and found that early (day 1 of life) elevations in several cytokines, adhesion molecules, and proteases were associated with an increased risk of developing BPD. Ambalavanan et al. (
34) analyzed serum from 1,067 preterm infants, with approximately 60% of them developing BPD. An early (<3 days of life) increase in levels of serum cytokines (both proinflammatory and anti-inflammatory) correlated with the development of BPD. The early detection of proinflammatory factors in these infants suggests that
in utero processes (e.g., chorioamnionitis, infection) associated with preterm labor and delivery initiate a fetal inflammatory response within the lung that continues after birth. All of these bioactive agents recruit and activate leukocytes, which can generate excessive ROS and cause significant pulmonary damage, including breakdown of capillary endothelial integrity and leakage of macromolecules (e.g., albumin) into alveolar spaces. Albumin leakage and pulmonary edema are known to inhibit surfactant function and have been postulated to be important factors in the development of BPD (
35).
As the cycle of injury continues with further production and accumulation of inflammatory mediators and ROS, significant injury to the lung can occur during a particularly critical period of rapid growth (i.e., the six divisions from 24 to 40 weeks of gestation). This amplification phenomenon can be further accentuated when there are inadequate concentrations and activity of anti-inflammatory agents such as IL-10 and clara (club) cell proteins (CC10) (
36,
37). Ramsay et al. (
36) demonstrated that reductions in expression and activity of CC10 in tracheal aspirates were
associated with an increased risk of death or the development of BPD in preterm infants. This suggests that this homeostatic protein that has potent anti-inflammatory properties may be essential in minimizing lung injury in high-risk infants. All of these studies clearly support the notion that this abnormal inflammatory process is a major contributing factor to the acute and the chronic changes in the lungs of infants with BPD.
Infection
Subclinical intrauterine infection and the ensuing inflammatory response have been clearly implicated in the etiology of preterm labor and premature rupture of membranes (
38). Significant basic science and epidemiologic evidence indicates that prenatal infection and inflammation are risk factors for the subsequent development of BPD, although not all studies are in complete agreement (
32). While several investigators have found a lower incidence of RDS in preterm infants born to mothers with chorioamnionitis (possibly due to an adaptive response to
in utero stress), they also observed a significantly higher rate of BPD in these infants (
38,
39). This suggests that although intrauterine infection may accelerate lung maturation, the ensuing inflammatory response may also “prime the lung,” causing progressive inflammation, lung injury, and subsequent inhibition of lung growth. Animal models have reinforced this concept with postnatal exposure to hyperoxia and mechanical ventilation further exacerbating this injury process (
40).
Ureaplasma urealyticum (UU) colonization in pregnant women has been implicated in the pathogenesis of preterm labor, premature rupture of membranes, and preterm delivery (
41). In addition, the organism has been detected (by culture and polymerase chain reaction) in tracheal aspirates from high-risk infants developing BPD suggesting that UU is an important factor in the pathogenesis of BPD (
32,
42). Viscardi and Hasday recently reviewed a significant number of conflicting studies examining the role of UU colonization (antenatal and postnatal) and suggested that antenatal UU infection in conjunction with postnatal causative factors (e.g., hyperoxia, mechanical ventilation) act as stimuli for a dysregulated inflammatory response, with recruitment of leukocytes and the production of cytokines and ROS ultimately leading to impaired alveolarization and the development of BPD (
41).
Normal defense mechanisms against infection can be compromised in the preterm lung, especially when infants are intubated and exposed to multiple courses of broad spectrum antibiotics. This makes them more susceptible to colonization and subsequent infection with a variety of infectious agents (e.g., virus, bacteria, fungi) that may increase the incidence and severity of BPD (
32). Several large clinical studies have found a strong correlation between lateonset sepsis and the development of BPD, usually with organisms such as staphylococcus epidermidis (
43,
44). These infections are associated with increased morbidity, mortality, and length of hospital stay and appear to contribute to the incidence and severity of BPD.
Nutrition
The nutritional status of the critically ill premature infant may also be important in the development of BPD. Adequate calories and essential nutrients for growth may be lacking during a period of stress and growth; vital components for immunologic and antioxidant defenses may be inadequate; and the nutritional supplements provided may actually contribute to ongoing damage. Premature infants have increased nutritional requirements because of increased metabolic needs and rapid growth requirements. If these increased energy and protein needs are not met, the infant will develop a catabolic state (e.g., negative nitrogen balance), which likely contributes to the pathogenesis of BPD. Inadequate nutrition interferes with normal growth and maturation of the lung and may potentiate the deleterious effects of oxygen and mechanical ventilation. Newborn rats fed a low-calorie diet have decreased lung weights, protein levels, and DNA content (
45). These abnormalities are even greater in pups that are nutritionally deprived at birth and exposed to hyperoxia.
Antioxidant enzymes may play a vital role in the protection of the lung and the prevention of BPD. Many of these enzymes have trace elements (e.g., copper, zinc, selenium) that are an integral part of their structure. Deficiencies in these elements may compromise the premature infant’s defenses and predispose the lung to further injury. The repair of elastin and collagen is limited in animals that are undernourished, and copper and zinc may be necessary for this repair (
46). While supplementation with these elements may provide protection to the lung and prevent hyperoxic lung injury, clinical trials using limited dosing strategies have not been able to prevent BPD (
47). Vitamin deficiency has also been postulated to be important in the development of BPD. While current nursery feeding and hyperalimentation regimens appear to provide adequate amounts of vitamin E for preterm infants, a relative decrease in the concentrations of other vitamins may be important in the pathogenesis of BPD. For example, Vyas et al. (
48) reported significant decreases in levels of ascorbate (which can function as an antioxidant) in tracheal aspirate fluid (TAF) from infants developing BPD compared to control infants, suggesting that supplementation with vitamin C might be beneficial. However, early supplementation with high-dose “antioxidant” vitamins has been studied in preterm baboons and was not efficacious in preventing lung damage from prolonged hyperoxia (
49).
Concentrations of vitamin A (i.e., retinol) may also be deficient in very-low-birth-weight infants (
50,
51). This vitamin appears to be important in maintaining cell integrity and in tissue repair with deficiency associated with changes in the ciliated epithelium of the tracheobronchial tree (
52). Shenai et al. (
50) demonstrated lower plasma retinol concentrations in the first month of life in infants who subsequently developed BPD. Despite adequate supplementation, some infants remain vitamin A deficient, presumably from increased absorption of parenteral vitamin A into the tubing of the intravenous administration set or from higher nutritional requirements (
53). A multicenter trial of vitamin A supplementation in premature infants at risk for developing BPD demonstrated that large doses of intramuscular vitamin A given three times per week was associated with a small (7%), but significant reduction in the incidence of BPD (
6). However, follow-up of these infants to 1 year CGA failed to demonstrate any significant improvement in clinical pulmonary status of vitamin A treatment, which has diminished the enthusiasm for this therapy (
7).
Large volumes of intravenous fluids are often administered to premature infants to provide adequate amounts (secondary to increased insensible water losses) and sufficient nutrition. Excessive fluid administration can be associated with the development of a patent ductus arteriosus (PDA) and pulmonary edema, which can lead to an increase in oxygen and ventilator requirements and the subsequent risk of BPD (
54,
55). Although early closure of the PDA using indomethacin or surgical ligation has been associated with improvements in pulmonary function, these approaches have not substantially affected the incidence of BPD and potentially have worsened outcome, leading many scientists and clinicians to question the validity of aggressive treatment of PDA (
56).
Genetics
A significant number of studies have been conducted in order to establish if genetic susceptibility is associated with an increased incidence and severity of BPD. Early studies observed that neonates were more likely to develop BPD if there was a strong family history of atopy and asthma. Nickerson and Taussig (
57) found a positive family history of asthma in 77% of infants with RDS who subsequently developed BPD, compared with only 33% who did not. Other investigators have supported the concept that strong genetic and environmental links do exist in infants who develop BPD (
32). Lavoie et al. (
58) studied 318 preterm twins of known zygosity who
were ≤30 weeks of gestation and found that heritability factors contributed significantly to the development of BPD. These data suggest that genetic markers may be useful for stratifying risk and targeting interventions to prevent BPD. Other investigators have conducted genome-wide association studies on independent groups of preterm infants ≤28 weeks of gestation or analyzed single nucleotide polymorphisms (SNPs) and have identified specific genes that appear to be associated with an increased risk for developing BPD (
59,
60). Finally, Cohen et al. (
61) performed expression cord profiling in umbilical cord tissue and demonstrated that infants who developed BPD had distinct chromatin remodeling and histone acetylation pathways, suggesting that epigenetic influences are critically important in the pathogenesis of BPD. As significant advances occur in analyzing large amounts of genetic information, further delineation of risk factors in larger populations will be needed to more definitively identify infants at high risk of developing BPD (
62).