Brain Tumors and Spinal Tumors
Kenneth J. Cohen and Ian F. Pollack
Central nervous system (CNS) neoplasms are, as a group, the most common solid tumors of childhood, second only to the leukemias as a cause of cancer in children. Despite improvements in diagnosis and management, more children will die of brain tumors than of any other type of pediatric cancer.1 Although the prognosis for children with certain CNS tumors, such as medulloblastoma,2 has been improved through a combination of surgical advances and refinements in radiotherapy and chemotherapy, other groups, such as diffuse intrinsic brainstem gliomas,3 continue to have a poor outcome. Children who experience long-term survival after therapy are at risk for sequelae from the tumor or its treatment that may adversely impact their quality of life.4 Current cooperative group studies are attempting to address these issues, focusing on improving survival results, with treatment-responsive lesions, in children with tumor types that historically have been resistant to therapy, as well as on improving quality of life in children with tumor-responsive lesions. Increasingly, these studies also incorporate molecular and biological classification of tumors to facilitate risk-adapted treatment stratification.
 EPIDEMIOLOGY AND RISK FACTORS
EPIDEMIOLOGY AND RISK FACTORS
Brain tumors occur at an incidence of 3.1 per 100,000 children per year. There is no gender bias, with a male-to-female ratio of 1.2:1.0 in Caucasian children and 0.9:1.0 in African American children. 
Genetic Causes of Brain Tumors
A variety of familial syndromes and diseases have been identified that predispose the patient to a variety of brain and other tumors. The most common of these disorders is neurofibromatosis 1 (von Recklinghausen disease). Additional disorders include neurofibromatosis 2, tuberous sclerosis complex (TSC), Von Hippel-Lindau (VHL) disease, Cowden disease, Li-Fraumeni syndrome, Turcot syndrome, and Gorlin syndrome. Summary information on each of these syndromes is detailed in eTable 460.1. 
Environmental Factors
Numerous studies have attempted to evaluate a variety of environmental exposures as possible causes for the development of childhood brain tumors. To date, only ionizing radiation exposure has been definitely proven to increase the risk of brain tumors. Data from the Children’s Cancer Study Group of over 6000 children who received radiation for leukemia demonstrated a 21.7-fold increase in CNS neoplasms, most predominantly high-grade astrocytomas. 
Many other exposures have been evaluated, including pesticides, tobacco, infectious agents, trauma, parental occupation, medications, electromagnetic fields, N-nitroso compounds, and vitamins. In most cases, risks appear modest, and follow-up studies often reach different conclusions.
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
The presenting signs and symptoms of children with brain tumors are quite variable and are impacted by numerous factors, including the size, rate of growth, and location of the tumor; the presence or absence of increased intracranial pressure; the tendency of the tumor to infiltrate normal brain structures; and the age of the child. Tumors may present in any of the following ways:
1. Nonlocalizing findings
2. Increased intracranial pressure
3. Localizing findings
Nonlocalizing findings include signs and symptoms such as headache, behavioral changes, developmental delay, weight gain/loss, and others. Tumors that present in this way are often located in noneloquent (nonfunctional) areas of the brain. These tumors are often challenging to diagnose because symptoms overlap with many common pediatric conditions, and if the tumor is slow growing, a lengthy period from the onset of symptoms to eventual diagnosis is the norm.
Increased intracranial pressure (ICP) is most frequently caused by obstructive hydrocephalus but can also occur when large tumors cause direct mass effect. In some cases of tumors with diffuse dissemination, increased ICP may be caused by communicating hydrocephalus. The classic symptoms of raised ICP include headache (irritability in infants) and vomiting. Both the location and the intensity of the headache can vary, and morning headaches, which improve during the day, are often described. Vomiting may or may not be associated with nausea. For very young children, a slowly progressive rise in ICP may manifest as a bulging fontanelle or separation of cranial sutures. Other findings may include lethargy, and in school-aged children, declining academic performance. Signs associated with increased ICP may include papilledema, Parinaud syndrome, cranial nerve palsies (particularly 6th nerve palsies), and neck pain in the setting of tonsillar herniation. Other signs (eg, anisocoria, ataxia, head tilt) can present and may reflect direct compression by the tumor or transmitted pressure on normal brain structures.
Localizing signs are, by definition, dependent on tumor location. When the diagnosis of a brain tumor is being considered in a child, it should be recognized that the majority of tumors in children are infratentorial (see Table 460-1). This contrasts with tumors in adults, which are predominantly supratentorial. The one exception is children under 1 year of age, in which there are equal numbers of infratentorial and supratentorial presentations. The infratentorial compartment includes the posterior fossa, the brain stem, and the region of the fourth ventricle (eFig. 460.1  ). Given the propensity for tumors to arise in this location in children, localizing symptoms often include cranial neuropathies (eg, 6th and 7th nerve palsies), ataxia, and long-tract signs. Other localizing signs include hemiparesis (including early handedness or change in handedness), hemisensory loss, vision disturbances, and seizures.
). Given the propensity for tumors to arise in this location in children, localizing symptoms often include cranial neuropathies (eg, 6th and 7th nerve palsies), ataxia, and long-tract signs. Other localizing signs include hemiparesis (including early handedness or change in handedness), hemisensory loss, vision disturbances, and seizures.
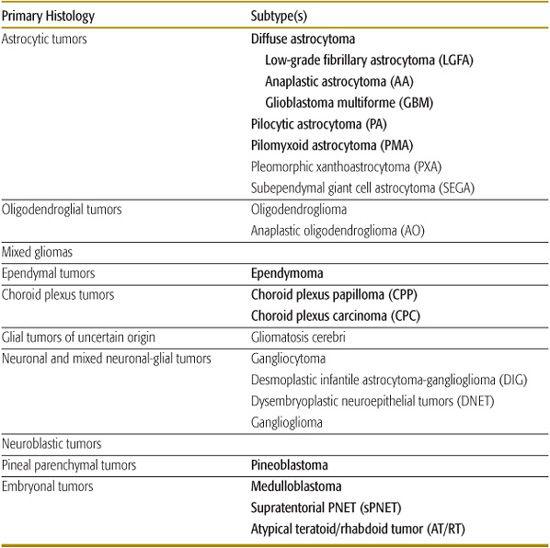
Table 460-1. Histologic Classification of Common Pediatric Brain Tumors
 DIAGNOSIS
DIAGNOSIS 
MRI remains the gold standard for diagnostic imaging. Diagnosis by MRI must include imaging before and after the administration of gadolinium and should include the full complement of T1 and T2 sequences. Advantages of MRI over CT images include markedly improved anatomic detail, the ability to identify small, noncontrast-enhancing lesions and the presence of leptomeningeal disease, and the absence of bony artifacts, which compromise imaging particularly of the posterior fossa by CT scan. 
 GENERAL PRINCIPLES OF MANAGEMENT
GENERAL PRINCIPLES OF MANAGEMENT
The mainstay of treatment for most brain tumors has relied on multimodality therapy with surgery, radiation therapy, and chemotherapeutics. The utility of each of these modalities is dependent on a variety of issues, including the location of the tumor within the brain or spinal cord, the age of the child, and the predicted chemosensitivity of the particular tumor.
Neurosurgical Therapy
Surgery typically comprises the first component in brain tumor management because this provides a mechanism for establishing the histological diagnosis and, in some cases, for reducing the tumor burden. Specific exceptions include tumors that have a diagnostic MRI appearance, such as diffuse brainstem gliomas and optic-hypothalamic tumors in children with neurofibromatosis-1, which are inherently unresectable and are appropriately treated with nonsurgical approaches based on an imaging diagnosis alone. 
Radiation Therapy
Radiation therapy (XRT) is potentially useful in the treatment of most pediatric brain tumors whether malignant or benign. The widespread application of XRT is limited by the age of the patient, the total area of brain requiring treatment, and the prior dose and distribution of XRT when retreatment is considered. In general, the younger the child, the greater are the efforts to reduce or eliminate XRT when feasible. 
Chemotherapy and Other Agents
The role of chemotherapeutics in the treatment of children with brain tumors is in evolution. Historically, chemotherapy was used in the adjuvant setting and often reserved for cases in which surgical resection and radiation therapy had failed to adequately control the tumor. Certain tumors have shown remarkable sensitivity to a variety of chemotherapeutics, which has led to the increasing use of these agents as a component of primary treatment. In addition, chemotherapy has been widely utilized in the management of the youngest children with brain tumors in an effort to avoid altogether, or delay, the eventual application of radiotherapy. 
COMMON PEDIATRIC BRAIN TUMORS
The World Health Organization (WHO) subdivides brain tumors into the following broad categories:
1. Tumors of neuroepithelial tissue
2. Tumors of peripheral nerves
3. Tumors of the meninges
4. Lymphomas and hematopoietic neoplasms
5. Germ cell tumors
6. Tumors of the sellar region
7. Metastatic tumors
With the exception of germ cell tumors and tumors of the sellar region, almost all other pediatric brain tumors are derived from tumors of neuroepithelial tissue. The WHO divides that category into many subgroups, with those seen in children listed in Table 460-1 (tumors in italics are discussed in this chapter). In addition, germ cell tumors and the tumor of the sellar region, craniopharyngioma, are discussed in some detail.
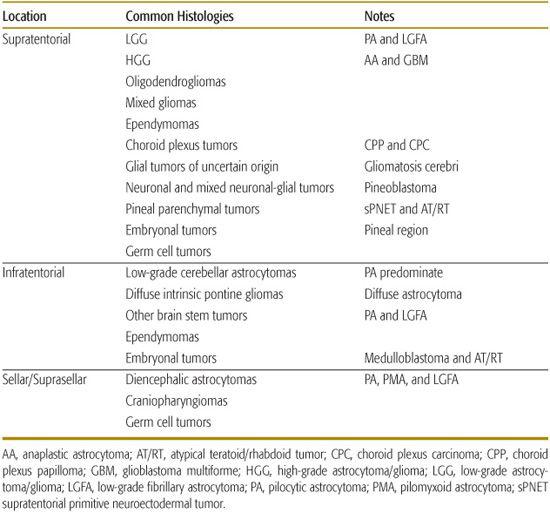
Tumors can be classified as supratentorial, infratentorial, and sellar/suprasellar. As mentioned, with the exception of infants, the majority of children will present with tumors in the infratentorial compartment. The locations of common pediatric tumors are listed in Table 460-2.
Table 460-2. Location within the Brain of Common Pediatric Brain Tumors
LOW-GRADE GLIOMA
Low-grade glioma (LGG) is a general term which encompasses the World Health Organization (WHO) grade 1 tumors (pilocytic astrocytoma and other histologically low-grade tumors) and the WHO grade 2 tumors (low-grade fibrillary astrocytoma and more recently pilomyxoid astrocytoma). In total, LGG constitute the largest group of childhood brain tumors. Pilocytic astrocytomas are the most common of these tumors in children (eFig. 460.2  ), representing approximately 25% of all childhood brain tumors. Prognosis is most dependent on the extent of tumor removal. Patients in whom a gross total resection has been achieved have a greater than 90% long-term survival.
), representing approximately 25% of all childhood brain tumors. Prognosis is most dependent on the extent of tumor removal. Patients in whom a gross total resection has been achieved have a greater than 90% long-term survival. 
For those children in whom surgical complete excision is deemed neither feasible nor safe, alternative treatments are considered. For selected children, expectant observation of a very slow-growing lesion might be recommended. When further treatment is required, both chemotherapy and radiation therapy can be utilized. 
HIGH-GRADE GLIOMA
The WHO uses the term high-grade glioma to encompass anaplastic astrocytomas (WHO 3) and glioblastoma multiforme (WHO 4). High-grade gliomas are the most common intrinsic malignant brain tumor in adults but are comparatively uncommon in childhood, accounting for less than 10% of tumors in most regions of the brain, with the exception of the brainstem (discussed in the next section). As with comparable lesions in adults, the prognosis for children with these tumors continues to be poor.8-10 Patients with grade IV lesions (ie, glioblastoma multiforme) have a worse prognosis than those with anaplastic astrocytoma or other grade III lesions and those with lesions not amenable to extensive re-section (eFig. 460.3  ), typically tumors that are located deep in the brain or are highly infiltrative, have a worse prognosis than those with more superficial, circumscribed, resectable lesions.
), typically tumors that are located deep in the brain or are highly infiltrative, have a worse prognosis than those with more superficial, circumscribed, resectable lesions. 
In view of the discouraging results with several preradiation and postradiation chemotherapy regimens for these tumors, ongoing studies are examining the efficacy of administering chemotherapy concurrently with irradiation, with the goal of potentiating the effects of both modalities, followed by additional therapy with these agents after irradiation. 
BRAINSTEM GLIOMA
Brainstem gliomas can be broadly characterized into focal and diffuse lesions. Focal tumors are commonly low-grade histologically and reasonably well circumscribed and include such subgroups as dorsally exophytic brainstem gliomas and focal lesions of the midbrain, medulla, and cervicomedullary junction,21,22 which have a comparatively favorable prognosis after treatment with surgery alone or combined with adjuvant therapy. These lesions characteristically present with slowly progressive focal neurological symptoms, such as facial weakness or eye movement palsies, or, for exophytic lesions, with symptoms relating to obstructive hydrocephalus, such as headache, nausea, and vomiting. The widespread availability of MRI has helped to distinguish such tumors from the larger group of diffuse brainstem gliomas, which are biologically aggressive and highly infiltrative malignancies (eFigs. 460.4 and 460.5  ). The latter tumors most commonly affect children younger than 10 years of age and characteristically present with rapidly progressive cranial neuropathies and long-tract signs, such as ataxia and weakness. As with other deep-seated, unresectable high-grade gliomas, the prognosis for children with these lesions remains poor, with long-term survival rates below 10%.
). The latter tumors most commonly affect children younger than 10 years of age and characteristically present with rapidly progressive cranial neuropathies and long-tract signs, such as ataxia and weakness. As with other deep-seated, unresectable high-grade gliomas, the prognosis for children with these lesions remains poor, with long-term survival rates below 10%.
To date, irradiation is the only modality that has been shown to consistently induce therapeutic responses, albeit transiently, in patients with diffuse brainstem tumors. 
EPENDYMOMA
Numerous institutional and cooperative group studies have observed that the most important predictor of survival among patients with ependymomas is the extent of tumor removal. Whereas children with tumors who undergo radiologically complete resection have a 50% to 75% chance of long-term survival after postoperative irradiation, less than 30% of those who have subtotal resections experience prolonged survival.27-29 Anaplastic tumor histology has also been noted to have an adverse association with outcome in several studies.27,30 Tumor location is a third factor that has been associated with outcome, in part reflecting that nonanaplastic supratentorial lesions are often more amenable to complete removal than are their infratentorial counterparts (eFig. 460.6  ).
).
CHOROID PLEXUS TUMORS
Choroid plexus tumors include the benign choroid plexus papilloma and the more aggressive choroid plexus carcinoma (CPC). These tumors are quite rare with an estimated annual incidence of 0.3 cases per million and account for approximately 1% of all brain tumors. CPC occurs in 20% to 40% of all cases of choroid plexus tumors. Complete resection of choroid plexus papilloma (eFig. 460.7  ) is generally curative with 10-year survival rates of 85%.34 Chemotherapy and/or radiation therapy is routinely recommended for children with choroid plexus carcinoma in which the tumor has not been completely resected at diagnosis. In those rare cases when choroid plexus tumors disseminate, multimodality therapy is recommended, but prognosis is poor.
) is generally curative with 10-year survival rates of 85%.34 Chemotherapy and/or radiation therapy is routinely recommended for children with choroid plexus carcinoma in which the tumor has not been completely resected at diagnosis. In those rare cases when choroid plexus tumors disseminate, multimodality therapy is recommended, but prognosis is poor. 
MEDULLOBLASTOMAS, SUPRATENTORIAL PRIMITIVE NEUROECTODERMAL TUMORS, AND PINEOBLASTOMA
Primitive neuroectodermal tumors (PNETs), the largest group of childhood malignant brain tumors, include location-specific subgroups referred to as medulloblastoma, pineoblastoma, and supratentorial PNETs. Medulloblastomas occur with a median age of 7 to 8 years and exhibit a slight male predominance. Pineal and supratentorial PNETs occur with a somewhat younger age distribution than medulloblastomas. 
From a therapeutic perspective, studies in the 1980s and 1990s demonstrated that these tumors could be subdivided into average-risk and high-risk groups based on differences in outcome after treatment with standard doses of irradiation (approximately 3600 cGy to the craniospinal axis with a boost to a dose of 5400 cGy to the tumor bed).35-37 Average-risk or standard-risk PNETs included lesions of the posterior fossa that had been extensively resected, had no evidence of metastatic disease (ie, were M0), and occurred in children older than 3 years of age. Patients with these tumors had a 5-year progression-free survival rate in the range of 60%. In contrast, high-risk tumors that had extensive residual disease, metastases, arose outside the posterior fossa, or occurred in children younger than 3 years were associated with survival rates below 40%.35-37
Based on these clinical risk factors, subsequent studies have tailored treatment, seeking to intensify therapy in high-risk PNETs in the hope of improving survival and to refine therapy in the standard-risk group in an effort to diminish treatment-related sequelae.2,38,39 In standard-risk patients, this effort centers on strategies to reduce the doses of radiotherapy needed to maintain a high level of survival, while decreasing radiation-related cognitive and endocrine toxicity, and has incorporated adjuvant chemotherapy in conjunction with 2340 cGy of cranio-spinal irradiation and boost irradiation to the posterior fossa.2
In contrast to the overall approach to standard-risk medulloblastomas, which is exploring ways to maintain favorable outcome results while reducing treatment-related sequelae, the general strategy for high-risk PNETs has focused on improving the frequency with which children become long-term survivors.38,45,46 Recent studies have capitalized on the activity of both irradiation and chemotherapy for these tumors by combining chemotherapeutic agents with radiosensitizing properties, such as carboplatin, with standard-dose irradiation followed by additional postradiation chemotherapy.
ATYPICAL TERATOID/RHABDOID TUMOR
Atypical teratoid/rhabdoid tumor is a rare neoplasm seen predominantly in young children. It is estimated that 10% to 15% of all malignant brain tumors in children under 3 years of age are atypical teratoid/rhabdoid tumors.47 In addition to the common histologic finding of rhabdoid cells, the hallmark of the diagnosis is now demonstration of a deletion or mutation of the INI1 (hSNF5) gene on chromosome 22q11.2, which occurs in the majority of cases.48 Historically, the prognosis was quite poor, with most children rapidly dying of their disease. More recently, aggressive multimodality therapy with surgery, radiation therapy, and chemotherapy, similar to that provided to patients with sarcomas, appears to have improved the outlook and chances for long-term survival of children with these tumors. 
GERM CELL TUMORS
Central nervous system germ cell tumors are broadly divided into germinomas and nongerminomatous germ cell tumors (NG-GCT). In the West, the incidence of germ cell tumors is approximately 1% to 3%, which contrasts sharply with the incidence in the Far East, which is 5 to 8 times greater.  NG-GCT tumors include embryonal carcinoma, choriocarcinoma, yolk sac tumors, and mixed germ cell tumors. In contrast to germinomas, which generally require a biopsy for diagnosis, NG-GCTs can often be diagnosed by characteristic imaging findings along with associated elevation of cerebrospinal fluid α1-fetoprotein and/or β-HCG.49
NG-GCT tumors include embryonal carcinoma, choriocarcinoma, yolk sac tumors, and mixed germ cell tumors. In contrast to germinomas, which generally require a biopsy for diagnosis, NG-GCTs can often be diagnosed by characteristic imaging findings along with associated elevation of cerebrospinal fluid α1-fetoprotein and/or β-HCG.49
Presenting symptoms vary but often reflect the propensity for germ cell tumors to arise in the suprasellar or pineal region. 
Owing to the sensitivity of germ cell tumors to radiotherapy and chemotherapy, complete surgical resection at the time of initial diagnosis is often not required or recommended. Once a diagnosis is established by biopsy or elevated tumor markers, subsequent therapy can be provided, which in many cases will result in definitive cure of the tumor. Historically, radiation therapy alone was the mainstay of therapy and resulted in overall survival in the 60% to 70% range for patients with germinoma. CNS germ cell tumors are remarkably sensitive to chemotherapy, and small studies have demonstrated excellent long-term survival with chemotherapy when given in combination with radiation therapy. Multimodality therapy is routinely utilized for patients with NG-GCT, but despite this approach, overall survival is substantially worse than for patients with germinoma. 
CRANIOPHARYNGIOMA
Craniopharyngiomas arise in the suprasellar region (eFig. 460.11  ) in or adjacent to the pituitary stalk.50 Embryologically, they are thought to result either from neoplastic change of cellular remnants of the primordial craniopharyngeal duct or from squamous metaplasia of cells within the pars intermedia of the pituitary gland. These tumors generally manifest with a combination of endocrine deficits, in particular short stature, resulting from compression of the hypothalamic-pituitary axis; visual impairment secondary to optic nerve, tract, and chiasmal compression; and hydrocephalus secondary to obstruction of the third ventricle.51
) in or adjacent to the pituitary stalk.50 Embryologically, they are thought to result either from neoplastic change of cellular remnants of the primordial craniopharyngeal duct or from squamous metaplasia of cells within the pars intermedia of the pituitary gland. These tumors generally manifest with a combination of endocrine deficits, in particular short stature, resulting from compression of the hypothalamic-pituitary axis; visual impairment secondary to optic nerve, tract, and chiasmal compression; and hydrocephalus secondary to obstruction of the third ventricle.51
Historically, these tumors were treated at many centers by surgical resection.52 Although progression-free survival rates in excess of 70% have been reported with complete surgical resection as the sole therapeutic modality, recurrence rates are high in the setting of residual disease.50 Moreover, radical surgical removal carries risks of endocrine deficits and neurological impairment. Accordingly, some surgeons have favored the use of more limited resections coupled with adjuvant radiation, which seem to provide rates of disease control that are comparable to those achieved with complete resection.53
BRAIN TUMORS IN INFANTS
Because the infant brain is particularly susceptible to the adverse effects of irradiation, malignant brain tumors in children younger than 3 to 5 years of age have generally been treated with different therapeutic protocols than those in older children.56,57 The challenges in managing these lesions are magnified because their symptoms are often nonspecific, including macrocephaly, failure to thrive, and delay or loss of developmental milestones, and as a result, the tumors are often large at the time of diagnosis.  Because investigators have historically been reluctant to treat very young patients with radiotherapy, chemotherapy has formed a cornerstone in the postsurgical management. Although 30% to 40% of children will respond to initial chemotherapy and not require radiotherapy, most will develop progressive, ultimately fatal disease within 1 to 2 years of diagnosis.
Because investigators have historically been reluctant to treat very young patients with radiotherapy, chemotherapy has formed a cornerstone in the postsurgical management. Although 30% to 40% of children will respond to initial chemotherapy and not require radiotherapy, most will develop progressive, ultimately fatal disease within 1 to 2 years of diagnosis.
To improve these results, recent studies have examined whether intensifying chemotherapy by adding a second, consolidation regimen or administering focal irradiation to the tumor bed can increase the frequency with which patients maintain long-term disease control. Preliminary data suggest that survival percentages may be improved by these approaches. 
LONG-TERM IMPACT OF TREATMENT
The treatment of children with brain tumors has been influenced and complicated by the impact of the tumor, surgery, irradiation, and chemotherapy on the developing brain. There is often a delicate balancing act between application of “curative” therapy and the impact of that therapy on the quality of life of the child following treatment.
Neurologic Toxicity
From the moment of tumor development, the patient is at risk for permanent neurologic toxicity. Many long-term sequelae are caused by the initial presence of the tumor and damage to normal brain structures by direct invasion of those structures by the tumor and/or development of hydrocephalus. Surgery poses numerous risks, and the aggressiveness of any surgical procedure must be balanced against the risk to normal structures in the operative bed.
Radiation therapy can cause impact during the delivery of radiation (eg, radiation-induced tumor swelling), shortly following radiation (eg, somnolence syndrome), when particularly large doses of radiation are delivered (eg, radiation necrosis), or many years after irradiation (eg, large vessel thrombosis, second malignancy). 
Neurocognitive sequelae in many ways represent the major complication of brain tumor treatment in children. While irradiation is often implicated as the primary cause of this long-term injury, the impact of other factors, such as the original damage caused by the tumor, surgery, and chemotherapy, cannot be underestimated. As discussed in the section on brain tumors in infants, therapeutic strategies are specifically designed to minimize or eliminate the use of irradiation in the youngest children because exposure to radiation during this time frame invariably results in progressive neurocognitive decline. When radiation is utilized, multiple measures of neurocognitive function will be impacted, including a substantial reduction in intelligence quotient. 
CONCLUSION
Improvements in neuroimaging, refinements in surgical techniques, and advances in the delivery of adjuvant radiotherapy and chemotherapy have led to meaningful improvements in the prognosis of several types of childhood brain tumors, such as standard-risk and high-risk medulloblastoma. In addition, molecular techniques are increasingly being examined and implemented as a way to refine therapeutic stratification for a number of tumor subgroups and may help to identify new targets for controlling tumor types that have been resistant to conventional treatments. Notwithstanding these improvements, several subgroups of childhood brain tumors, such as diffuse intrinsic brainstem gliomas, have a poor prognosis, and among the more favorable-risk tumor groups, there is a strong need to identify strategies that can reduce sequelae of the tumor and its treatment. Enrollment of affected children on cooperative group trials is recommended in order to ensure access to state-of-the-art therapy and to facilitate further advances in the management of childhood CNS tumors.
REFERENCES
See references on DVD.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


