Metabolic Disorders of Bone
Bone biopsies may be used in the diagnosis of metabolic bone diseases in the pediatric population, such as rickets and renal osteodystrophy. Bone biopsy may also be helpful in the assessment of treatment response. Features such as cortical thickness and cortical porosity are evaluated histologically.9 Radiographic, genetic, and molecular studies are the modalities most often used in diagnosing inherited metabolic bone disorders, such as osteogenesis imperfecta and thanatophoric dysplasia.
BONE-FORMING TUMORS
Osteoma
Osteomas are benign mass-like lesions composed of cortical bone that arise most commonly in the frontal and nasal sinuses and the outer calvarial bones. They are not considered to be neoplastic and are not surgically removed unless they become symptomatic or are cosmetically problematic. Osteomas may be biopsied in order to rule out other lesions of bone. Plain films reveal a dense, ivory-like sclerotic mass attached to the bony cortex. Histomorphology of osteomas is similar to that of compact cortical bone, revealing a dense mass of mature lamellar bone with decreased marrow.10
Osteoid Osteoma and Osteoblastoma
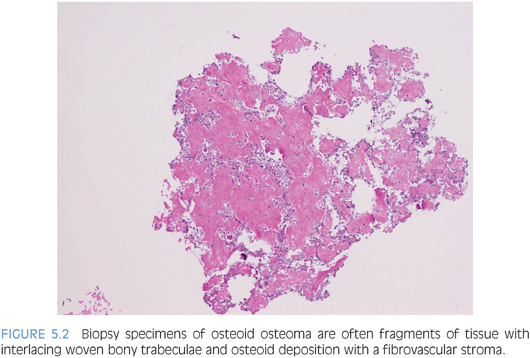
These are benign bone-forming tumors with a male predilection that present within the first two decades of life. Often, biopsy specimens for these two entities consist of fragments of curetted material. Histologically, osteoid osteoma and osteoblastoma are nearly identical with a few subtle differences. Location, size, and radiographic appearance of the lesion are helpful in differentiating between these entities. Osteoid osteoma comprises approximately 12% of all benign bone tumors,10 whereas osteoblastomas account for about 3%. Osteoid osteoma occurs most commonly in the long bones (70% to 80% of cases), especially the femur, tibia, and humerus,11 whereas osteoblastoma tends to arise in the axial skeleton, especially the vertebral column. Osteoid osteoma forms a discrete radiolucent bony nidus usually measuring less than 1 cm that is surrounded by sclerotic bone (eFig. 5.3). The classical clinical presentation is that of localized pain that worsens at night and is promptly relieved by salicylates or other nonsteroidal anti-inflammatory medications.10,11 Osteoblastoma may have a radiographic appearance similar to that of osteoid osteoma, although the nidus, if present, is larger (usually >2 cm) and more irregular in shape. Periosteal reactions exhibit much less reactive sclerosis than osteoid osteoma and the lesion may display an aggressive radiologic pattern.10
Sections of the nidus of osteoid osteoma and the lesional tissue of osteoblastoma reveal interlacing, irregular trabeculae of osteoid and woven bone (Fig. 5.2). Osteoblastic rimming of the trabeculae is a characteristic finding (eFig. 5.4), and the intervening fibrovascular stroma is rich in capillaries. Often in biopsy specimens, the osteoid osteoma or osteoblastoma fragments are interspersed among nonlesional bony tissue.
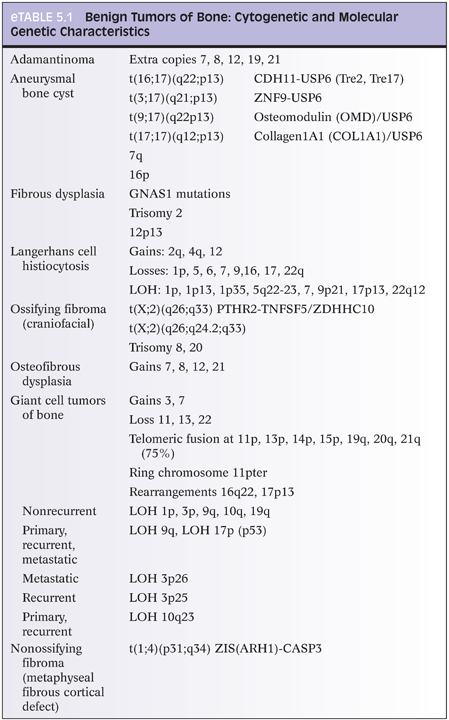
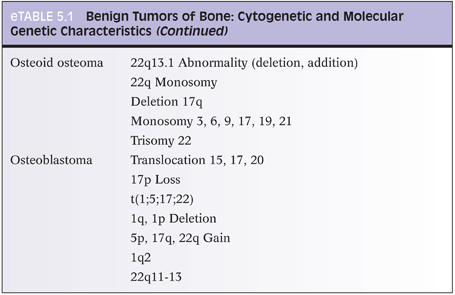
With osteoid osteoma, cytogenetic studies have shown recurrent chromosome 22 abnormalities (eTable 5.1).12 Certain genes located at 22q13 may play a role in osteoid osteoma development. With osteoblastoma, cytogenetic studies (eTable 5.2) have shown translocations with chromosomes 1, 5, 15, 17, 20, and 22. Loss of 17p (site of p53) has also been reported. Several chromosomes have deletions (1q, 1p), whereas others show gains (5p, 17q, 22q).

Surgical removal of the nidus is curative and results in immediate relief.11 Currently, osteoid osteoma lesions are often treated by interventional radiologists with thermal ablation therapy, with a biopsy being performed immediately before ablation. Confirmation of the diagnosis occurs after routine core needle biopsy processing, which may provide evidence of a nidus in some but not all cases. The differential diagnoses include stress fracture, bone island (enostosis), and low-grade osteosarcoma.10
Osteosarcoma
Osteosarcoma is the most common primary malignant bone tumor in children. It is an osteoid and/or bone-forming tumor that occurs most commonly in the rapidly growing regions of bone in adolescents, especially the distal femur and proximal tibia.13 Persistent bone pain is the usual presenting symptom. Risk factors include previous radiation exposure, genetic predisposition to the Li-Fraumeni, Bloom, or Rothmund-Thomson syndrome, and mutations in genes associated with osteosarcoma. However, the vast majority of osteosarcomas are sporadic. Tissue from the diagnostic biopsy should be submitted for cytogenetics and cryopreserved for possible molecular pathology analysis; in addition, touch preparations provide a means to perform FISH and CISH for specific gene loss and amplifications, as well as for detecting gene partner rearrangements in translocations that may occur in other tumors that mimic osteosarcoma, such as Ewing sarcoma.
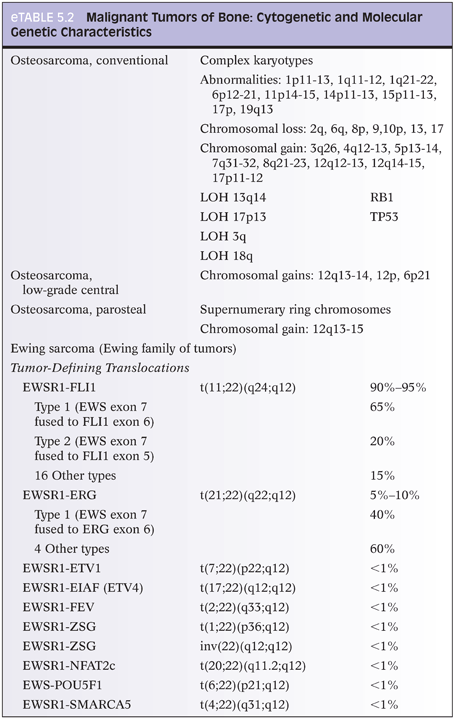
Osteosarcoma types all have the requisite histologic diagnostic feature of malignant osteoid, composed of malignant cells and associated osteoid matrix. Conventional osteosarcoma, the most common type, arises within the medullary cavity of bone and progresses to cortical penetration and soft tissue invasion. These tumors are high grade histologically and are characterized as osteoblastic, chondroblastic, or fibroblastic, depending on the predominant tumor matrix (Fig. 5.3). Small cell osteosarcoma is a rare variant that resembles Ewing sarcoma histologically and is composed of small round cells within a lacelike osteoid matrix. CD99 may be positive in these tumors, and exclusion of Ewing sarcoma/peripheral primitive neuroectodermal tumor (PPNET) by performing FISH, CISH, cytogenetics, and/or reverse transcriptase polymerase chain reaction (RT-PCR) is necessary to eliminate Ewing sarcoma/PPNET as a consideration.13 A rare but important type of osteosarcoma that must be differentiated from aneurysmal bone cyst (ABC) grossly and radiographically is telangiectatic osteosarcoma, which represents 3% of osteosarcomas. Telangiectatic osteosarcoma, like ABC, presents as a destructive, purely lytic lesion at the distal end of the bone. Fluid–fluid levels may be detected on magnetic resonance imaging (MRI).14 Unlike ABC, telangiectatic osteosarcoma has markedly pleomorphic, malignant, high-grade tumor cells, often with a malignant giant cell component (eFigs. 5.5 and 5.6). Low-grade central osteosarcoma is a rare variant (1% of osteosarcomas), characterized by low-grade cytomorphologic features and a mandibular or maxillary location. Periosteal osteosarcoma, parosteal osteosarcoma, and high-grade surface osteosarcoma are rare types of osteosarcoma that arise along the periosteal surface of the affected bone and together comprise 5% to 10% of osteosarcomas.
Radiographic features of conventional osteosarcomas are variable and reflect the location, extent, and predominant matrix component of the tumor. The lesion is usually poorly defined. Periosteal reactions are commonly seen in response to cortical penetration, typically in the form of a Codman triangle or a “sunburst” pattern (eFig. 5.7). Sclerosis extending into soft tissues is indicative of and specific for osteosarcoma.
The malignant osteoid-producing cells in osteosarcoma are atypical, pleomorphic, and hyperchromatic. Osteoid deposition is usually lacelike in conventional osteosarcomas. Trabecular bone formation is unusual, and osteoblastic rimming is not a feature. However, it is not unusual for osteosarcomas to use existing normal bony trabeculae as scaffolding for laying down neoplastic osteoid by tumor cells. The malignant osteoid often has a mosaic, wormian-like pattern with filling in of the marrow space. In rare cases of well-differentiated osteosarcomas, the osteoid material laid down between normal bony trabeculae may resemble reactive bone formation with the exception of malignant tumor cells seen within or at the periphery of the neoplastic osteoid. Numerous atypical mitoses are often seen. The nuclei may be small or inconspicuous in osteosclerotic areas. In telangiectatic and fibroblastic osteosarcoma, osteoid material may be extremely rare and require extensive searching and multiple tissue sections in order to identify osteoid and categorize the tumor as an osteosarcoma.
A large variety of karyotypic abnormalities have been described in osteosarcoma, including gain of regions of chromosome 1 and loss of regions of chromosomes 6, 9, 10, 13, and 17 (eTable 5.2).12 Mutations of Rb (13q14) and p53 (17p13) genes occur commonly in osteosarcoma, as well as abnormal expression of the c-jun, c-fos, c-myc, and H-ras oncogenes.13,17 The karyotype in osteosarcoma is characterized by numerous chromosomes (over 100) with duplications, loss of short and long arms of chromosomes, and loss of entire chromosomes, as well as insertions, inversions, and translocations. The complexity of the karyotype is usually reflected in the cytogenetic reports, and it is rare and unusual that a cytogenetic report will list all cytogenetic abnormalities. Simian virus 40 (SV40) has been detected in approximately one-third of osteosarcomas. This DNA polyomavirus inhibits p53 function, promoting cell proliferation and may play a role in oncogenesis in certain osteosarcomas.
Prognostic factors include tumor grade, location, type, tumor size, presence or absence of metastatic disease, complete resection with negative surgical margins, and posttherapy tumor necrosis (>90% tumor necrosis favorable).14 Current treatment of high-grade osteosarcoma consists of neoadjuvant chemotherapy followed by resection.15 Most often, limb-sparing procedures are performed. Amputation becomes necessary when inappropriate biopsy technique has been used that contaminates tissue planes, when pathologic fracture precludes limb salvage, and with progression of tumor during treatment that precludes limb salvage.
Differential diagnoses depend on the histologic type and include aggressive osteoblastoma (osteoblastic osteosarcoma), chondrosarcoma (chondroblastic osteosarcoma), ABC (telangiectatic osteosarcoma), and Ewing sarcoma (small cell osteosarcoma).
CARTILAGE-FORMING TUMORS
Osteochondroma
Osteochondroma (osteocartilaginous exostosis) is the most common bone tumor in children, comprising 58% of childhood bone tumors with a slight male predominance. Osteochondromas are exophytic bony lesions that may be pedunculated or sessile and are in continuity with the cortex of the underlying bone (eFig. 5.8). They may be found with any bone that undergoes endochondral ossification, including the scapula and iliac crest.13
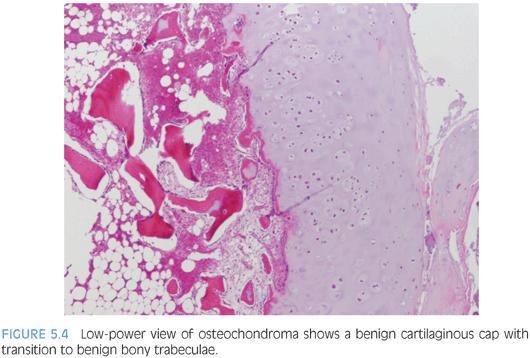
Osteochondromas vary in size from 1 to 2 cm in greatest dimension up to 15 to 20 cm and are covered by a cartilaginous cap that is gray-blue and glistening (Fig. 5.4). Histologically, the cartilaginous cap contains chondrocytes arranged in somewhat disorganized columns with a certain degree of chondrocyte cloning. Ossification occurs and mature trabecular bone is formed within the underlying stalk, similar to an epiphyseal plate.16 Occasional binucleated cartilaginous lacunae can be seen which is not a worrisome feature in young patients.14 Mutations in the EXT1 (8q24, Langer-Giedion syndrome), EXT2 (11p11-p12, DEFECT-11 syndrome), and EXT3 (19p) tumor suppressor genes involved in signaling pathways within the epiphysis have been implicated in multiple osteochondromatosis and associated syndromes (eTable 5.3).12

Chondrosarcomatous transformation is highly associated with a cartilage cap thickness of greater than 2 cm; in which case, additional or complete sampling of the tumor should be done. With cartilaginous cap thickness of between 1 and 2 cm, it is appropriate to diagnose these tumors as osteochondromas with atypical or borderline features and to recommend complete excision with follow-up. In general, all osteochondromas should undergo complete excision, which is curative. Close clinical follow-up for recurrence is indicated in those lesions with tumor at the resection margin. Multiple osteochondromas should be closely monitored radiographically for increasing size and thickness of the cartilaginous cap given their increased risk of sarcomatous transformation (up to 5% of cases).14,16
Enchondroma/Enchondromatosis
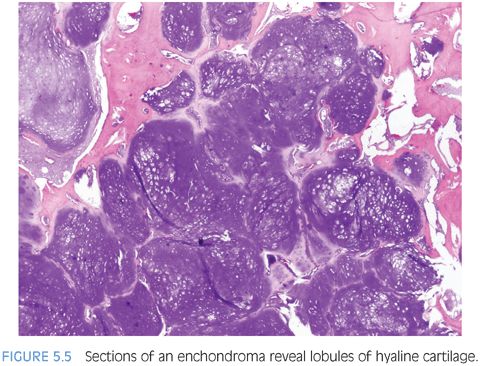
Enchondromas are benign tumors of mature hyaline cartilage that arise predominantly in the small tubular bones of the hand but can present in the large tubular bones. Enchondromas are the second most common bone tumor in children, representing 24% of all bone tumors in this age group.13 Clinical presentation is that of local pain and/or swelling or pathologic fracture in some cases.
Radiographically, enchondromas are well-defined, radiolucent lesions situated within the medullary cavity without cortical penetration. They may result in cortical expansion and thinning with endosteal scalloping. Stippled calcification within the enchondroma may be evident.13,16 Grossly, enchondromas are lobulated, glistening, and gray-blue. Microscopically, they resemble lobulated masses of mature, benign cartilage with variable cellularity (Fig. 5.5 and eFig. 5.9). Binucleated chondrocytes as well as increased cellularity may be seen, especially in the bones of the extremities in children.13 Several karyotypic abnormalities have been reported in Ollier disease, including deletion of 9p, which may indicate loss of CDKN2A tumor suppressor gene (eTable 5.4).12 Other common cytogenetic findings are trisomy 5, chromosome 15 extra copies, 12q13-15 rearrangement, and 6q rearrangement, which plays a role in chondrocyte differentiation (PTHrP, bcl-2).